expinput kallisto folder empty

Brian Estevez

Nathan Salomonis
--
You received this message because you are subscribed to the Google Groups "Alternative Splicing and Functional Prediction" group.
To unsubscribe from this group and stop receiving emails from it, send an email to alt_predictio...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/alt_predictions/3ca4b90f-cbc3-44be-abe5-ac7fd5041581o%40googlegroups.com.
Brian Estevez
On Thursday, July 2, 2020 at 2:02:57 AM UTC-4, Nathan Salomonis wrote:
Hi Brian,Can you send your log file from AltAnalyze and a screenshot of these directories? The default ICGS option for Minimum number of samples differing to 2 or 3 and correlation to 0.6 for this study design.Best,Nathan
On Thu, Jul 2, 2020 at 12:36 AM Brian Estevez <beste...@gmail.com> wrote:
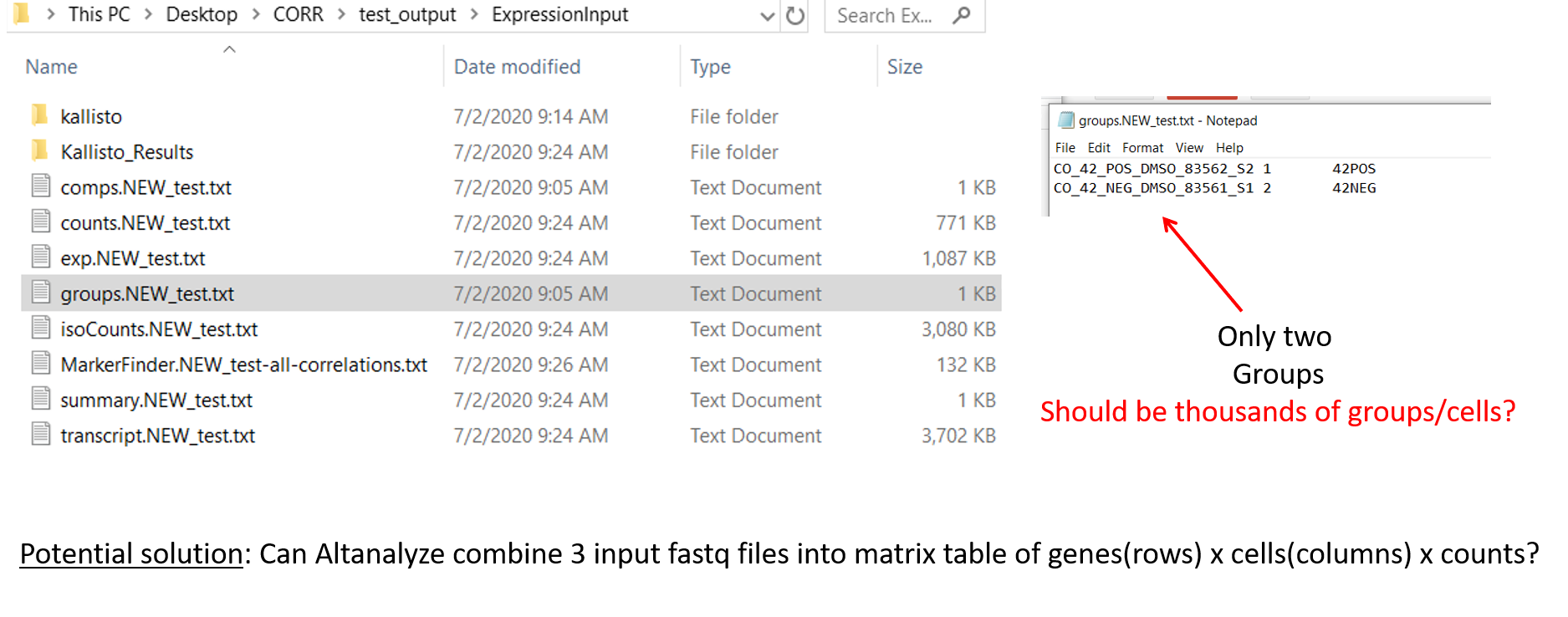
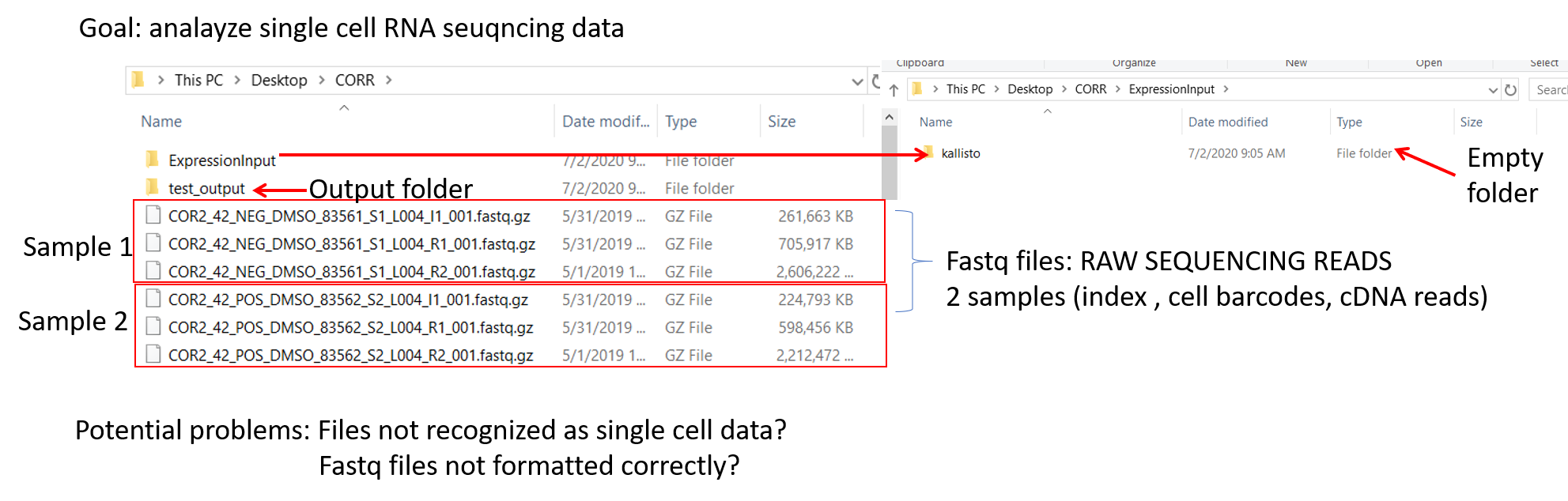
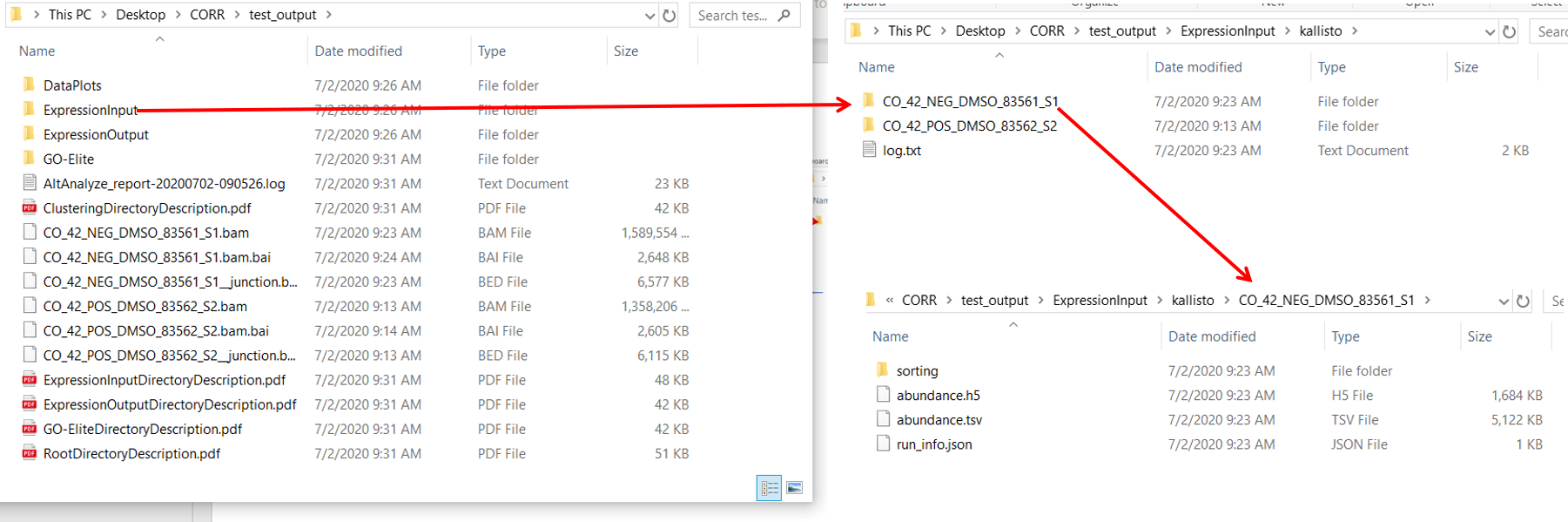
--Hi,I am using the latest version for PC 2.1.4.1My goal is to do ICGS on single cell rna sequencing fastq files. From GUI, I first run raw sequence or processed on 6 fastq files: from 2 samples, each with index, cdna and cell barcode files). I do this in order to get a Kalisto expression input file and re-run for ICGS using process expression file mode.Oddly, there are two expression input folders being generated upon completion of first run on my fastq files:1) in the specified output folder and another one 2) alongside the fastq files.There are 3 files in '1)' two folders that correspond to two of my samples containing files such as abundance.h5, abundance.tsv, and run_info.json, and a log.txt file.There are no files in ' 2)' ExpressionInput/KallistoSo I use the exp.txt file in Kalisto_Results folder. I run analysis and get the error: indexerror list inder out of range.What should I try next?-Brian
You received this message because you are subscribed to the Google Groups "Alternative Splicing and Functional Prediction" group.
To unsubscribe from this group and stop receiving emails from it, send an email to alt_pre...@googlegroups.com.
{kind=link}
{kind=link}
{kind=link}
Brian Estevez
On Thursday, July 2, 2020 at 2:02:57 AM UTC-4, Nathan Salomonis wrote:
Hi Brian,Can you send your log file from AltAnalyze and a screenshot of these directories? The default ICGS option for Minimum number of samples differing to 2 or 3 and correlation to 0.6 for this study design.Best,Nathan
On Thu, Jul 2, 2020 at 12:36 AM Brian Estevez <beste...@gmail.com> wrote:
--Hi,I am using the latest version for PC 2.1.4.1My goal is to do ICGS on single cell rna sequencing fastq files. From GUI, I first run raw sequence or processed on 6 fastq files: from 2 samples, each with index, cdna and cell barcode files). I do this in order to get a Kalisto expression input file and re-run for ICGS using process expression file mode.Oddly, there are two expression input folders being generated upon completion of first run on my fastq files:1) in the specified output folder and another one 2) alongside the fastq files.There are 3 files in '1)' two folders that correspond to two of my samples containing files such as abundance.h5, abundance.tsv, and run_info.json, and a log.txt file.There are no files in ' 2)' ExpressionInput/KallistoSo I use the exp.txt file in Kalisto_Results folder. I run analysis and get the error: indexerror list inder out of range.What should I try next?-Brian
You received this message because you are subscribed to the Google Groups "Alternative Splicing and Functional Prediction" group.
To unsubscribe from this group and stop receiving emails from it, send an email to alt_pre...@googlegroups.com.
Nathan Salomonis
python AltAnalyze.py --platform RNASeq --species Hs --restrictBy protein_coding --excludeCellCycle no --removeOutliers yes --row_method hopach --ChromiumSparseMatrix /Users/GitHub/tests/demo_data/10X/input/cancer.h5 --output Users/GitHub/tests/demo_data/10X/input --runICGS yes --expname cancer --downsample 2500
For a counts file
python AltAnalyze.py --platform RNASeq --species Hs --restrictBy protein_coding --excludeCellCycle no --removeOutliers yes --row_method hopach --ChromiumSparseMatrix /Users/GitHub/tests/demo_data/10X/input/cancer.h5 --output Users/GitHub/tests/demo_data/10X/input --runICGS yes --expname cancer --dataFormat counts
You could also run Kallisto bus to get the input files, which I believe is fairly straight forward. Kallisto-bus is on our timeline but was delayed.
Best,
Nathan
To unsubscribe from this group and stop receiving emails from it, send an email to alt_predictio...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/alt_predictions/fded842a-fe43-45f1-ab61-b71a357b9bb9o%40googlegroups.com.