Scantwo function output interpretation
44 views
Skip to first unread message

Rohan Richard
Feb 2, 2023, 9:45:56 AM2/2/23
to R/qtl discussion
Hello,
Thank you in advance for your reply,
I am using the function scantwo to assess epistasis between my QTL. It is the first time I do this and I am not familiar with the output.
I would like some help for the interpretation (I have already read the scantwo description function :https://rqtl.org/tutorials/new_summary_scantwo.pdf for your notice). What conclusion can I draw from this table? Which of the five LOD should I consider (lod.full, lodd.fv1, lod.int, lod.add or lod.av1?) for evidence of epistasis? Also what is the usual threshold at which an epistasis effect would be qualified as significant?
Thank you in advance for your reply,
Rohan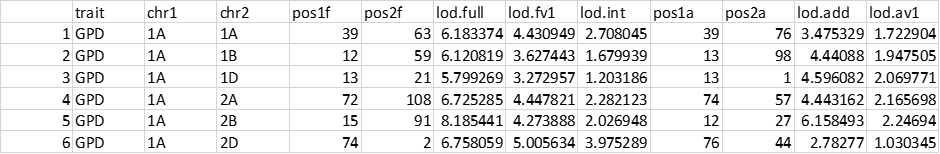

Karl Broman
Feb 2, 2023, 10:20:44 AM2/2/23
to R/qtl discussion
The best source of information about how to interpret the results of scantwo is chapter 7 of the R/qtl book.
Also see step 18 (pages 7-8) of the "Brief tour of R/qtl".
We look for lod.full to be large and either lod.fv1 or lod.int to also be large. lod.fv1 is the difference between the best two-locus epistatic model and the best single-locus model, while lod.int is the difference between the best two-locus epistatic model and the best two-locus additive model.
How large? It depends on the data, and particularly the type of cross and the size of the genome. There's unfortunately no good substitute for a scantwo permutation test, though it can be extremely time consuming, computationally. It also can be quite memory intensive. If Haley-Knott regression is appropriate for your data, use the function scantwopermhk(), which avoids some memory problems.
karl
Rohan Richard
Feb 3, 2023, 6:03:05 AM2/3/23
to R/qtl discussion
Good morning Karl,
Thank you very much for your reply!
Unfortunately, due to computation time I can't run the permutation tests on my machine which is too slow.
Do you have any idea about "good" LOD thresholds (lod.full, lod.fv1 and lod.int) for a DH population of wheat with 111 lines genotyped with 1459 markers ?
Best regards,
Rohan
Karl Broman
Feb 3, 2023, 8:05:18 AM2/3/23
to R/qtl discussion
I don’t; sorry
karl

Martin Ferris
Feb 3, 2023, 9:39:23 AM2/3/23
to rqtl...@googlegroups.com
I've found, to decrease the computational time, I can decrease my marker density to save on computational space. For example, I'll go from a 3000 marker map (with a 1,500 cM genome length) for initial scans and pare it down so that I only keep markers if they are great than/equal to 5 cM apart. That gave me a ~400 marker map. It made those permutations possible.
Regards,
Marty
--
You received this message because you are subscribed to the Google Groups "R/qtl discussion" group.
To unsubscribe from this group and stop receiving emails from it, send an email to rqtl-disc+...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/rqtl-disc/1c9c006c-4ab2-45c0-953b-12f96a32c9ban%40googlegroups.com.
Reply all
Reply to author
Forward
0 new messages