How to deal with large gaps in genetic map?
207 views
Skip to first unread message

Lara
Jun 1, 2022, 4:51:16 PM6/1/22
to R/qtl discussion
Hi Dr. Broman and R/QTL community,
I am concerned with the large gaps in my genetic map. I have a few chromosomes that are >1,000 cM and a several that are >200 cM. If it was a single or small group of markers, then I would think to drop them. However, there are distinct groups of markers with sizable distance between them and a decent number of markers in each group. What is the best way to proceed to reduce the distances on these chromosomes?
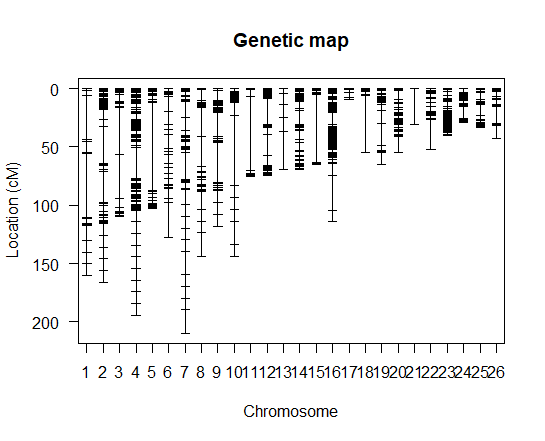
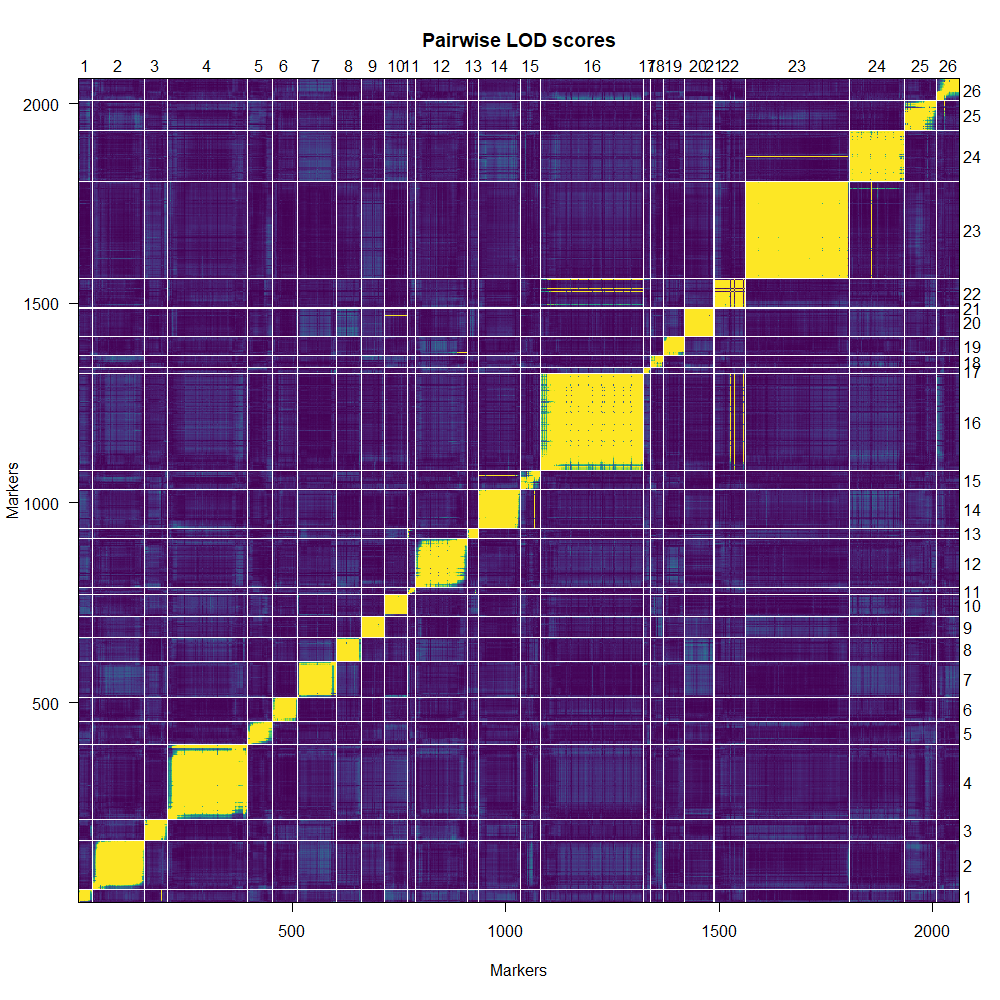
Background: Until this point, I used physical distance in cM and the linkage groups determined from alignment to a reference genome. Most markers have segregation distortion so I used general likelihood (markerlrt) instead of recombination fraction (est.rf) to assess their associations. Based on the RF plot, I dropped all markers that were linked only to themselves and not the rest of the chromosome. The RF plot (Chilling RF plot om.png) and genetic map (Geneticmap_chilling.om2.png) after dropping these markers is attached. I used droponemarker and dropped all markers with LOD>0. Then I re-estimated the map with est.map, and now some chromosomes are >1000 cM (Geneticmap_dropone1.png).

Karl Broman
Jun 1, 2022, 10:30:34 PM6/1/22
to R/qtl discussion
The large gaps occur because a marker or markers is not tightly linked to the surrounding markers.
So first figure out the markers that are in-between those big gaps, and then look at the relationship between their genotypes and the genotypes at the flanking markers. I'd be asking: Should I just omit these individual markers, or is there some other problem with the genotypes which can be fixed?
The iplotMap() function in the R/qtlcharts package can be useful, for highlighting the problem markers. Or just plot individual chromosomes with the marker names shown, and you might be able to read them off.
karl

{kind=link}
{kind=link}
{kind=link}
Martin Ferris
Jun 2, 2022, 1:39:49 PM6/2/22
to R/qtl discussion
I'm a little confused how you can get markers that are ~1000 cM apart. Conceptually, isn't 50 cM the maximum you could assess? Is it displaying them that way because qtl(2) is told they're on the same chromosome, so the +1000 cM distance spacing is an explicit flag in the program to let the user know they have an error in their map assumptions?
Message has been deleted
Karl Broman
Jun 2, 2022, 1:55:16 PM6/2/22
to R/qtl discussion
As the estimated recombination fraction approaches 1/2, the estimated genetic distance approaches infinity.
For the Haldane map function, 50 cM corresponds to a recombination fraction of 0.316.
qtl:::mf.h(50)
In estimating the genetic map with est.map(), R/qtl takes the marker order as fixed and known, estimates the recombination fractions between each pair of adjacent markers, and then converts those to cM distances with a map function. But recombination fractions aren't allowed to be too close to 0.5, so there's a maximum estimated distance that will be returned, I think 0.5 - 1e-14, which gives 1577 cM with the Haldane map function.
karl
Lara
Jun 2, 2022, 2:10:20 PM6/2/22
to R/qtl discussion
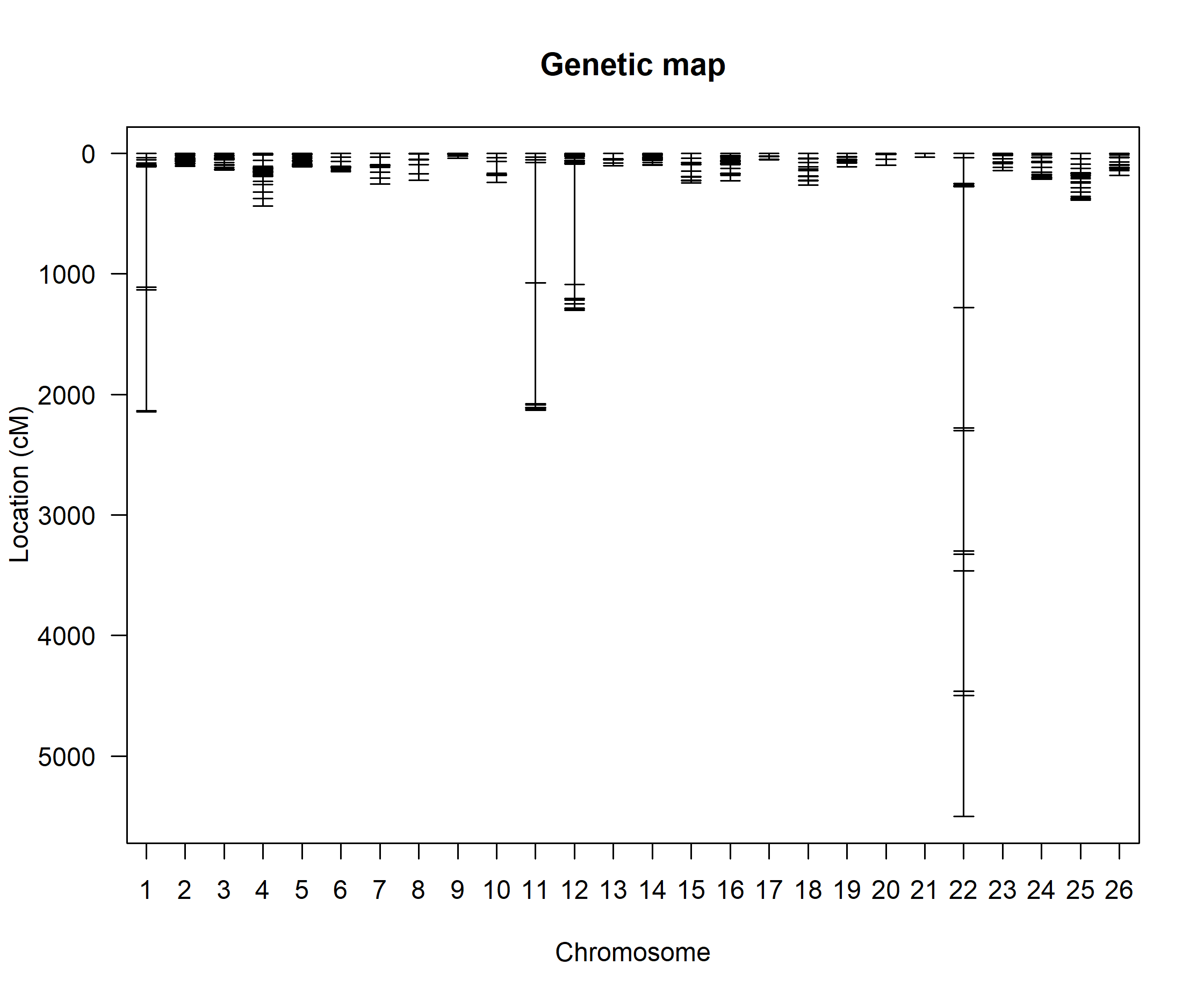
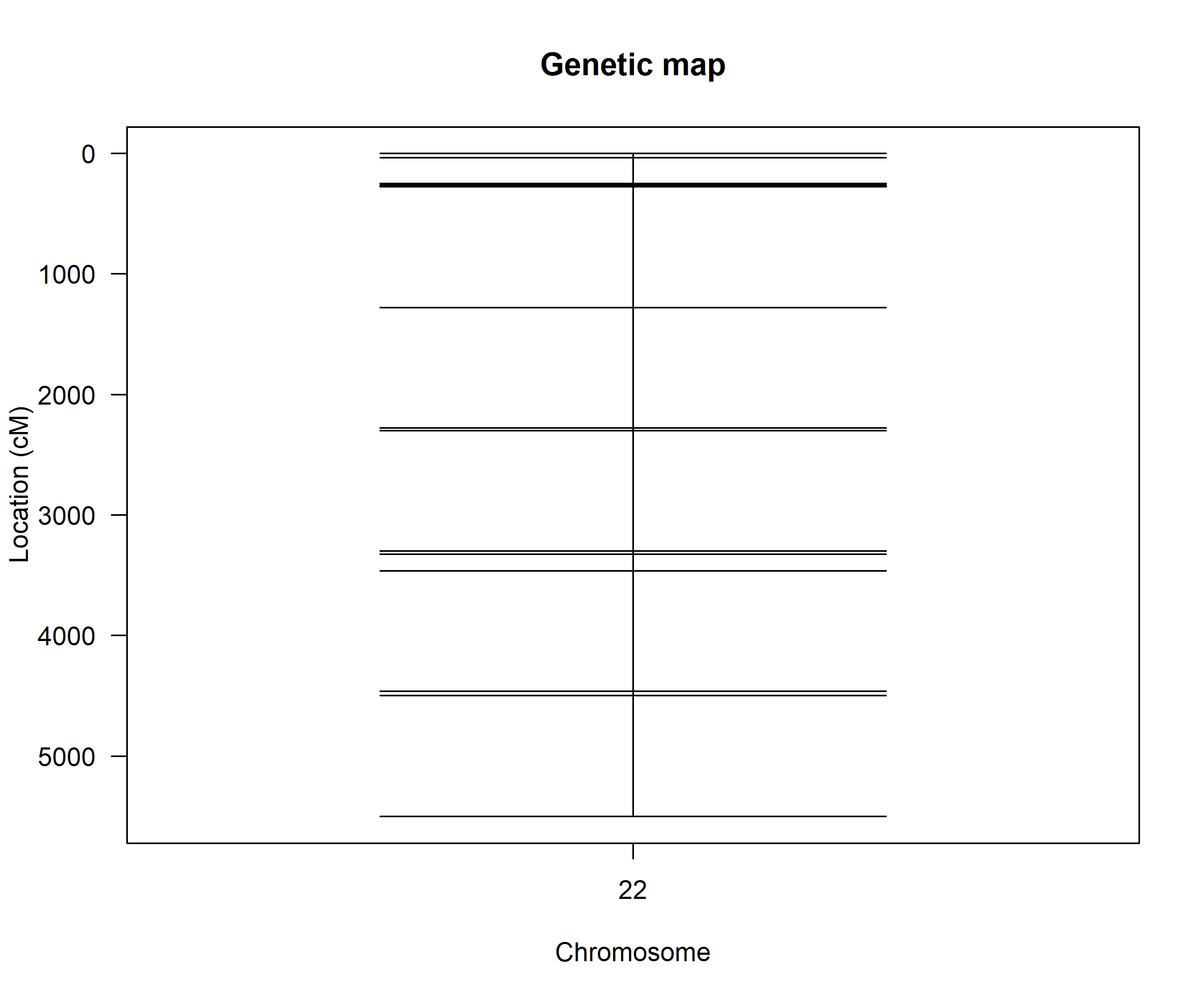
Dr. Broman, thanks that package looks really helpful! I'll try that on the chromosomes that have one or a few markers in between the groups. However, what is the best approach when all the markers are spaced out relatively evenly and there's no clear culprit marker to investigate (chr22 attached) or the markers "dumbbell" (chr12 attached)?
{kind=link}
{kind=link}
Karl Broman
Jun 2, 2022, 2:25:28 PM6/2/22
to R/qtl discussion
For each of the big gaps, I would look at the relationship between the genotypes at the marker on one side and the marker on the other.
karl
Lara
Jun 27, 2022, 4:16:35 PM6/27/22
to R/qtl discussion
Thank you. I removed the unlinked markers based on the RF map and the markers in the middle of the dumbbells on the genetic map, re-estimated the genetic map, repeated, and now it's looking much better! The largest chromosomes are now 250 cM instead of >2,000 cM, so it's on the right track!
Reply all
Reply to author
Forward
0 new messages