Best Way to Identify Multiple QTL in F1 (Outbred, 4-way Phase Known)
305 views
Skip to first unread message

Joshua Havill
Mar 13, 2021, 12:46:46 PM3/13/21
to R/qtl discussion
Hi Karl et al.
Following the various tutorials, I came to realize that 4-way crosses were limited to standard interval mapping (CIM and MQM won't work with 4-way crosses).
I detect what appears to be several QTL on a single linkage group but I'm having trouble trying to decipher the next steps to control for the 1 major QTL to decipher whether the other QTL are still significant.
Would the best approach be to use the scantwo() to identify interacting loci?
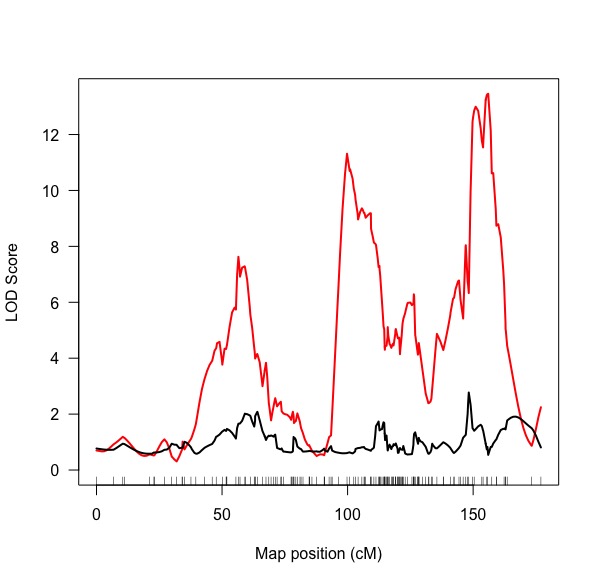
Pasted below is a picture from the scanone() output (in red) and the addpair() output using the original QTL as the formula (in black). I'm unsure though whether this was the appropriate approach though.
Cheers,
Josh

Karl Broman
Mar 14, 2021, 4:28:39 PM3/14/21
to R/qtl discussion
I think makeqtl/fitqtl/addqtl/etc work for “4way” crosses, as well as stepwiseqtl().
karl
havi...@umn.edu
Mar 14, 2021, 5:35:18 PM3/14/21
to rqtl...@googlegroups.com
Hi Karl,
I guess my question then is, what is the appropriate method for using addqtl()?
I fit a singular qtl using fitqtl() for the largest peak, then restricted the addqtl() to only LG 4 where the QTL was detected using that QTL and the formula as “binary ~ Q1”.
If the goal was to detect additional QTL, the manual describes leaving out the QTL of interest on the restricted chromosome but an error is thrown because no QTL is included.
-------
Joshua Havill
On Mar 14, 2021, at 3:28 PM, Karl Broman <kbr...@gmail.com> wrote:
I think makeqtl/fitqtl/addqtl/etc work for “4way” crosses, as well as stepwiseqtl().
--
You received this message because you are subscribed to the Google Groups "R/qtl discussion" group.
To unsubscribe from this group and stop receiving emails from it, send an email to rqtl-disc+...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/rqtl-disc/3af75de6-5424-4e38-9bf3-b117e3547b57n%40googlegroups.com.
Joshua Havill
Mar 15, 2021, 12:30:02 AM3/15/21
to R/qtl discussion
Hi Karl,
As a follow-up, I re-ran the analysis with scantwo() using a small number of permutations restricted to the linkage group that originally exhibited multiple QTL.
The output suggests there are two QTL, but I want to make sure that I am interpreting the output correctly. There is strong evidence for two two significant QTL, with non-significant interactions between loci, but a demonstrable additive effect of genotype on the phenotype? There also appears to be evidence for epistatic effects between loci on the phenotype of interest?
summary(pm.scan, perms = perm.a, pvalue = T)
pos1f pos2f lod.full pval lod.fv1 pval lod.int pval pos1a pos2a lod.add pval lod.av1 pval
com_04:com_04 100 156 24.2 0 10.7 0 3.52 1 100 156 20.7 0 7.16 0
pos1f pos2f lod.full pval lod.fv1 pval lod.int pval pos1a pos2a lod.add pval lod.av1 pval
com_04:com_04 100 156 24.2 0 10.7 0 3.52 1 100 156 20.7 0 7.16 0
If this is the case, is the plot for the plotLodProfile the correct plot to use to demonstrate that the third "QTL" is spurious or do I just expand upon the analysis in the text to describe the subsequent analyses taken to tease apart these relationships?
Thanks,
Josh
Karl Broman
Mar 15, 2021, 10:41:18 AM3/15/21
to R/qtl discussion
"If this is the case, is the plot for the plotLodProfile the correct
plot to use to demonstrate that the third "QTL" is spurious or do I
just expand upon the analysis in the text to describe the subsequent
analyses taken to tease apart these relationships?"
I would use makeqtl() to create a qtl object with the two QTL on the chromosome, and then addqtl() to look for evidence to add a third QTL.
karl
Joshua Havill
Mar 15, 2021, 2:23:13 PM3/15/21
to rqtl...@googlegroups.com
Hi Karl,
Can you specify how you would write the model to search for additional QTL after fitting just a single one using fitqtl() with the largest QTl effect fitted first?
qtl <- makeqtl(cross, chr = c(LG), pos = c(POS), what = c("prob"))
fit <- fitqtl(cross, pheno.col = "binary", qtl, formula = binary ~ Q1, get.ests = TRUE, method = "hk", model = "binary")
fit <- fitqtl(cross, pheno.col = "binary", qtl, formula = binary ~ Q1, get.ests = TRUE, method = "hk", model = "binary")
add <- addqtl(cross, pheno.col = "binary", qtl = qtl, formula = binary ~ Q1, method = "hk", model = "binary")
Thanks,
Josh
To view this discussion on the web visit https://groups.google.com/d/msgid/rqtl-disc/e20eb409-de19-4ed9-b0b5-2a20a3f82c59n%40googlegroups.com.
--
Joshua Havill, M. Sc.
C: (612)-836-7834
C: (612)-836-7834
Graduate Research Assistant, Ph. D. Candidate
Plant and Microbial Biology Graduate Program
Department of Agronomy and Plant Genetics
411 Borlaug Hall
1991 Upper Buford Circle
Saint Paul, MN 55108
University of Minnesota - Twin Cities
Karl Broman
Mar 15, 2021, 4:30:35 PM3/15/21
to R/qtl discussion
Take a look at the tutorial on multiple-QTL models, https://rqtl.org/tutorials/new_multiqtl.pdf
In particular, the section on addqtl starting on page 6.
Alternatively, look at chapter 9 of the R/qtl book, and particularly section 9.3.4 starting on page 267.
In that example, the call to addqtl looks like this:
out.aq <- addqtl(hyper, qtl=rqtl, formula=y~Q1+Q2+Q3*Q4)
The rqtl object has 4 QTL, and we're considering a model where the 3rd and 4th QTLs interact; that's why the formula is included.
The result of addqtl() will be similar to results of scanone(), but at each position we fit a five QTL model: the four QTL in the rqtl object
(with the 3rd and 4th interacting) plus an additional QTL at sliding position across the genome.
karl
Joshua Havill
Mar 16, 2021, 2:10:25 PM3/16/21
to R/qtl discussion
Hi Karl,
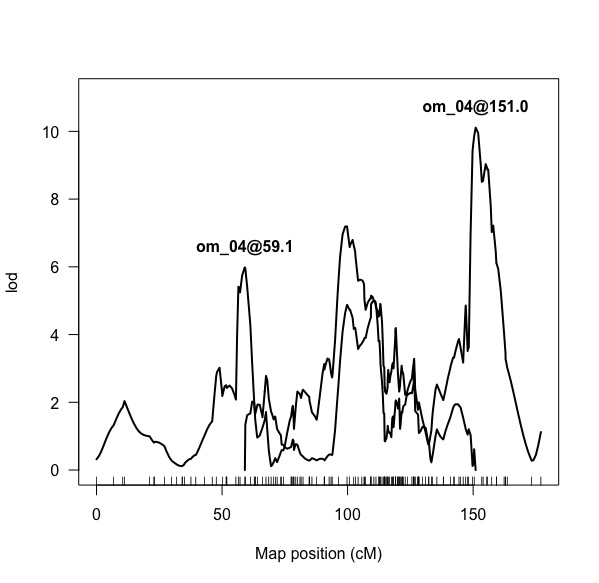
I've figured out the multiple QTL part with regards to specifying the model, etc. but now I'm trying to interpret the resulting LOD profile after (using the plotLodProfile()). These results are supported by scanone() and scantwo() results.
Reply all
Reply to author
Forward
0 new messages