how to separate more than two peaks in a chromosome?
146 views
Skip to first unread message

ziming zhong
Jul 2, 2018, 3:53:45 PM7/2/18
to R/qtl discussion
Hello!
I'm following the r/qtl book to analyse my QTLs. I got 3 peaks respectively in 3 chromosomes, as shown in the attached picture 1, one of which on chromosome 8 is with huge confidence interval. So I think there may be more than one genes in that peak and I want to use methods for multiple QTLs to separate them.
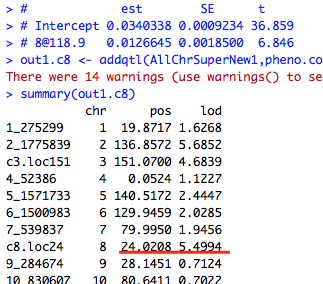
I first ran scaneone, and got the results as shown in attached picture 2. Then I controlled the QTL on chr8 by running makeqtl and addqtl of position 8@118, and the result showed me that there may be another peak at 8@24 (as shown in attached picture 3). So I expected that if I control the QTLS on chr2 and chr3, then I will find two peaks seperately at 8@120 and 8@24 on chr8.
But the problem came. After I ran add pair command, the result gave me two peaks at 8@75 and 8@100 with very weak evidence (shown in attached picture 4). So I don't understand why the result is so different from what I expected according to the results from previous steps.
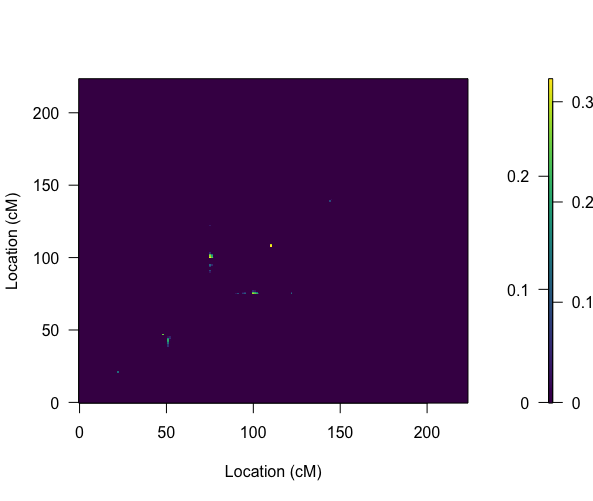
Btw, when I plot the addpair result, I got the graph as shown in attached picture 5, indicating three locations interacting in chr8. But the evidence is very weak.
Could somebody explain a bit for me? Are there more than one gene in the peak on chr8? If so, did I do the right thing to separate them? Thanks in advance!
Here is my code:
out1.bin <- scanone(AllChrSuperNew1, pheno.col=2, method=c("em"))
summary(out1.bin)
qtl8 <- makeqtl(AllChrSuperNew1,chr = 8, pos =118.8633 ,what="prob")
summary(fitqtl(AllChrSuperNew1,pheno.col=2,method = "hk",qtl=qtl8,get.ests = TRUE))
out1.c8 <- addqtl(AllChrSuperNew1,pheno.col=2,qtl=qtl8)
summary(out1.c8)
plot(out1.c8,ylab="LOD Score")
qtl23 <- makeqtl(AllChrSuperNew1,chr= c(2,3),pos=c(136.8572,151.0700),what="prob")
out1.ap <- addpair(AllChrSuperNew1,qtl=qtl23,chr=8,method = "hk",verbose=FALSE)
summary(out1.ap)

Karl Broman
Jul 2, 2018, 11:25:11 PM7/2/18
to rqtl...@googlegroups.com
It looks like you’re approaching this correctly. In my view, the 3 peaks you see in the initial LOD curve on chr 8 are not strong indications for multiple QTL on the chromosome. The multiple-QTL analyses you’ve done, which provide only weak evidence for additional QTL, suggest to me that those initial bumps where just bumps and not additional QTL.
Karl
--
You received this message because you are subscribed to the Google Groups "R/qtl discussion" group.
To unsubscribe from this group and stop receiving emails from it, send an email to rqtl-disc+...@googlegroups.com.
To post to this group, send email to rqtl...@googlegroups.com.
Visit this group at https://groups.google.com/group/rqtl-disc.
For more options, visit https://groups.google.com/d/optout.
<2_3_8.pdf>
<Screen Shot 2018-07-02 at 21.24.46.png>
<Screen Shot 2018-07-02 at 21.25.05.png>
<Screen Shot 2018-07-02 at 21.25.25.png>
<Rplot copy.png>
ziming zhong
Jul 3, 2018, 5:27:30 AM7/3/18
to R/qtl discussion
Hello Karl! Thanks for your quick reply!
Yeah, I agree that evidence for a second qtl on chr8 is quite weak. But what i don't understand here is that if the second qtl is weak, then why I get the a QTL with LOD=5 at 8@24 after I control the QTL on chr8 (after addqtl step)? I mean if the second qtl on chr8 is truly weak, should I just get a weak LOD for chr8 after I control the qtl on it? I'm kind of confused here.
ziming zhong
Jul 3, 2018, 9:51:19 AM7/3/18
to R/qtl discussion
Hello Karl!
Sorry that I have another question: if there is only one qtl in chr8, why do you think nearly the whole chromosome have such high LOD score? Does that indicate my markers are bad? I got several QTL cases like this that the qtl peaks have very wide and flat top and large numbers of markers in the entire chromosome have very high LOD score. What do you think could be the reason for this? Is there any way for me to reduce my confidence interval?
Thanks a lot!
On Tuesday, July 3, 2018 at 5:25:11 AM UTC+2, Karl Broman wrote:

{kind=link}
{kind=link}
{kind=link}
{kind=link}
benc...@gmail.com
Feb 23, 2021, 4:41:54 PM2/23/21
to R/qtl discussion
What does your linkage map look like?
Reply all
Reply to author
Forward
0 new messages