Filter for significant phenotypes
51 views
Skip to first unread message

Lena Flörl
Feb 7, 2023, 5:24:12 PM2/7/23
to R/qtl discussion
Hello again!
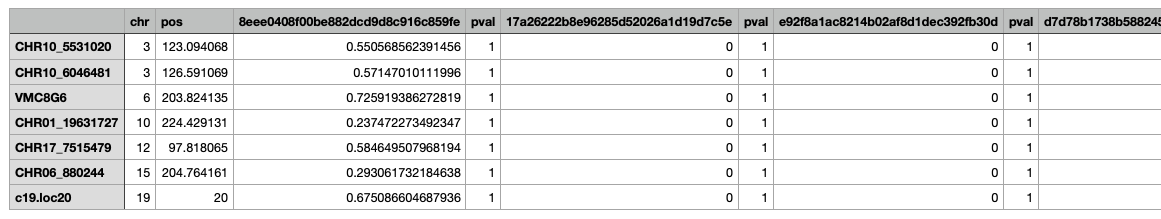
I am running r/qtl with multiple phenotypes (>1000 at a time) but my problem is that when I try to summarise all peaks above the LOD threshold and with a significant p-value, I get a huge data frame containing all traits and their values at the respective marker position
This is the code I use:
summary(out.bac, format="allpheno", perms=operm.bac, alpha=0.05, pvalues=TRUE)
And the output dataframe looks like this (but with thousands of columns..):
Does anyone know how to filter this data frame in R to only contain the significant traits and their LOD scores and p-values? Or maybe there is a trick to using "summary"?
So far I could only filter by p-value but I do not know how to retain the prior column containing the trait ID and LOD score.
Thanks a lot! :)
Lena

Karl Broman
Feb 7, 2023, 6:29:33 PM2/7/23
to R/qtl discussion
With either format="allpheno" you'll end up with 2000 columns, because it will include the LOD score and p-value for each trait on each chromosome. With format="allpeaks", you'll end up with 3000 columns, as it will give the peak location, LOD score, and p-value for each trait.
You're maybe more interested in format="tabByChr" or format="tabByCol" which will then give a list of data frames (one per chromosome or one per trait) with the set of peaks for that chromosome or trait.
karl
Lena Flörl
Feb 7, 2023, 7:40:25 PM2/7/23
to R/qtl discussion
Hi Karl,
thanks for the super quick reply - "tabByCol" is exactly what I needed!
Maybe one last, quick question: do you also have a suggestion on how to extract the data then?
Because I cannot export it as a csv file or even transform to a dataframe anymore when it's grouped by trait. For example:
>> Error in (function (..., row.names = NULL, check.rows = FALSE, check.names = TRUE, :
arguments imply differing number of rows: 0, 1, 2Thanks so much!
Lena
Karl Broman
Feb 7, 2023, 10:56:20 PM2/7/23
to R/qtl discussion
The output of summary with format="tabByCol" is a list of data frames, and so you would need to combine them.
Here's an example, using the hyper data, to create the summary for 1000 synthetic phenotypes:
set.seed(20230207) # to make this reproducible
data(hyper)
hyper <- calc.genoprob(hyper)
hyper <- calc.genoprob(hyper)
n_phe <- 1000
hyper$pheno <- matrix(rnorm(nind(hyper)*n_phe), ncol=n_phe)
colnames(hyper$pheno) <- paste0("phe", 1:n_phe)
out <- scanone(hyper, pheno=1:n_phe, method="hk")
operm <- scanone(hyper, pheno=1, method="hk", n.perm=1000)
out_sum <- summary(out, perms=operm, alpha=0.2, format="tabByCol")
operm <- scanone(hyper, pheno=1, method="hk", n.perm=1000)
out_sum <- summary(out, perms=operm, alpha=0.2, format="tabByCol")
Most of the elements of out_sum have no rows; let's just keep the ones that are not empty
out_sum <- out_sum[ sapply(out_sum, nrow) > 0 ]
length(out_sum) # 219
For each table, let's add a column that gives the phenotype name and then make the third column just "lod".
Let's also put the marker names (row names) into a column.
for(i in seq_along(out_sum)) {
out_sum[[i]] <- cbind(out_sum[[i]],
marker=rownames(out_sum[[i]]),
phenotype=names(out_sum)[i])
}
Now we paste them all together into a single data frame. I don't really understand how this works to be honest.
out_combined <- do.call("rbind", out_sum)
The output is a data frame
head(out_combined)
# chr pos ci.low ci.high lod marker phenotype
# phe1 4 74.3 47.0 74.3 2.455963 D4Mit14 phe1
# phe10.D1Mit132 1 43.7 35.0 64.5 3.393348 D1Mit132 phe10
# phe10.D5Mit99 5 73.2 32.8 82.0 2.392715 D5Mit99 phe10
# phe15 16 0.0 0.0 25.1 2.363224 D16Mit32 phe15
# phe16 7 55.6 13.1 55.6 2.141917 D7Nds4 phe16
# phe17 11 25.1 13.1 80.9 2.166632 D11Mit310 phe17
# phe1 4 74.3 47.0 74.3 2.455963 D4Mit14 phe1
# phe10.D1Mit132 1 43.7 35.0 64.5 3.393348 D1Mit132 phe10
# phe10.D5Mit99 5 73.2 32.8 82.0 2.392715 D5Mit99 phe10
# phe15 16 0.0 0.0 25.1 2.363224 D16Mit32 phe15
# phe16 7 55.6 13.1 55.6 2.141917 D7Nds4 phe16
# phe17 11 25.1 13.1 80.9 2.166632 D11Mit310 phe17
Lena Flörl
Feb 7, 2023, 11:16:20 PM2/7/23
to R/qtl discussion
Hi Karl,
I cannot thank you enough! This amazingly helpful!
Thanks so much!
Cheers!
Lena
Lena Flörl
Feb 15, 2023, 7:51:04 PM2/15/23
to R/qtl discussion
Hi Karl,
working with these exported markers I just noticed that some of them have names that are not in my input files. My guess is that r/qtl is creating these names maybe when I'm using Jittermap? I can imagine that the significant marker 'c17.loc260' was inserted at 260 cM on linkage group 17?
Is there a way to get the next (real) marker in such cases?
Thanks a lot!
Best,
Lena
Karl Broman
Feb 15, 2023, 10:01:40 PM2/15/23
to R/qtl discussion
Those are “pseudomarkers”. If you just want to do calculations at the markers, use step=0 in the call to calc.genoprob.
To find the nearest marker, you can use the function find.marker().
karl
Lena Flörl
Feb 16, 2023, 3:53:16 PM2/16/23
to R/qtl discussion
Hi Karl - thanks a lot!
Reply all
Reply to author
Forward
0 new messages