Z-coordinate Issue and Loading Multiple Runs Into a Single Imaging File
114 views
Skip to first unread message

Dmitry Leontyev
Feb 4, 2022, 4:47:28 PM2/4/22
to Cardinal MSI Help
Hello everyone,
I am new to Cardinal and have ran into some issues that I am hoping to get some more information on.
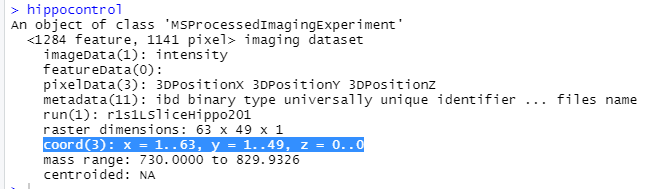
1) After reading in my .imzML file using readMSIData(), when I type the name of my imaging data set and hit enter, this is the output I get:
Notice how in the pixelData row there are 3 coordinates and if you look further you can see "coord(3): x = 1..63, y = 1..49, z = 0..0". When I looked at the practice data sets they did not have a z coordinate. I think the z coordinate going from 0 to 0 might lead to empty pixel problems. I am simply exporting my data as a .imzML file from Bruker SCiLS lab and then loading it in. I even tried to make a new .imzML file and the same thing happened. Any thoughts on this?
2) I image my control and treated tissues separately, so they are in different .imzML files. I read them in individually and so I will have one control data set and one treated data set, with only one run in each of the data sets. But then I encounter issues with running things like PLS because in the examples a single image data set is simulated with three runs and crossvalidation is called on the data set with all three runs at once (whereas in my case I have two data sets with one run each). So my question is how are we supposed to read in multiple .imzML files into individual runs within a single data set? Essentially how should I read in my control and treated .imzML files as separate runs into one imaging data set?
Thank you,
Dmitry

Gordon Luu
Feb 4, 2022, 4:58:17 PM2/4/22
to Cardinal MSI Help
1. This has to do with how SCiLS exports imzML files. It includes the Z dimension, and since everything is on the same plane (i.e. z=0), you see z=0..0 in cardinal since that entire column is == 0. If you are using a Bruker instrument, exporting from flexImaging doesn't include this and has worked better for me personally than exporting from SCiLS.
2. Cardinal has a combine function (Cardinal::combine()) that you can use to combine separate runs. To run statistical analysis, each condition should have 3 runs (3 biological and/or technical replicates, preferably biological) for best results.
- Gordon
Dmitry Leontyev
Feb 7, 2022, 12:54:58 PM2/7/22
to Cardinal MSI Help
Godron,

I greatly appreciate the input. Excuse me for the delayed response, your response notification went into my junk folder.
When I have tried to combine runs using cbind(), I get the error below. Do you think this might be due to different dimensions of the files? When reading the manual on combining it mentions that it works "assuming certain conditions are met",
"The m/z-values must match between all datasets to successfully combine them" and "Some processing may be necessary to ensure datasets are compatible before combining them". What steps do you take to ensure that datasets are compatible?
"The m/z-values must match between all datasets to successfully combine them" and "Some processing may be necessary to ensure datasets are compatible before combining them". What steps do you take to ensure that datasets are compatible?
Dmitry Leontyev
Feb 7, 2022, 1:51:23 PM2/7/22
to Cardinal MSI Help
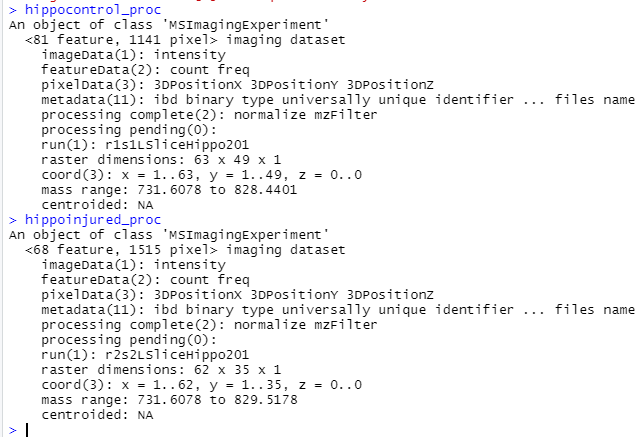
Here is information on the two data sets I was trying to use cbind() on. Seems as if the number of features, the number of pixels and the mass range between the data sets is not the same.
On Friday, February 4, 2022 at 4:58:17 PM UTC-5 go...@ucsc.edu wrote:

educh...@gmail.com
Feb 7, 2022, 2:15:47 PM2/7/22
to Cardinal MSI Help
Hi Dmitry,
If you want to combine two datasets in one, you need to do the binning processing step.
This steps allow you to get the same m/z for each of them if you use the same binning.
Try to do it and tell us if this work for you
Best regards,
Edu
Dmitry Leontyev
Feb 7, 2022, 3:29:44 PM2/7/22
to Cardinal MSI Help
When exporting from flexImaging, do you tend to choose the "Export Spectra from Disk" option or the "Export from flexImaging (processed)" option?
On Friday, February 4, 2022 at 4:58:17 PM UTC-5 go...@ucsc.edu wrote:
Dmitry Leontyev
Feb 7, 2022, 4:10:46 PM2/7/22
to Cardinal MSI Help
Hi Edu,
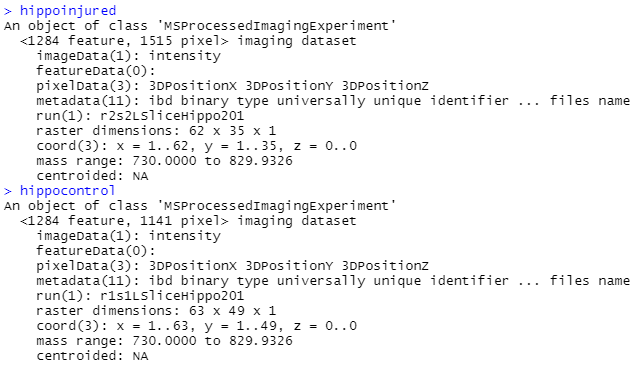
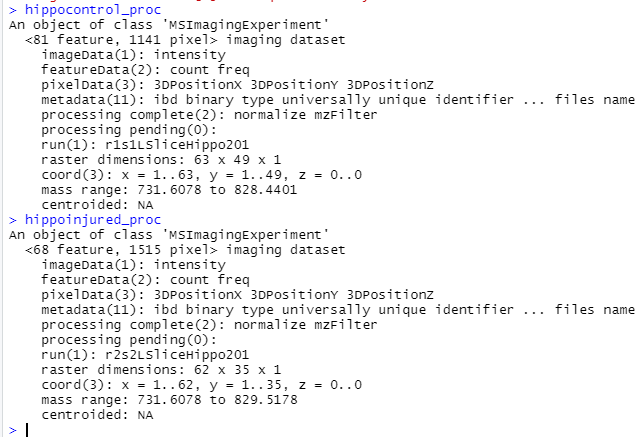
I am binning when I am individually loading in my control and injured image files (code below), but I think because I am processing them individually that they dont end up with the same m/z range, so I can't combine them (but I also don't know how to process them together). I tried to add the binning step to pre-processing code and the mass ranges still did not match. The outputs of hippocontrol, hippoinjured, hippocontrol_proc and hippoinjured_proc are included below the code. You can notice that before processing, hippocontrol and hippoinjured have the same mass range, but after processing they do not. Perhaps this is because of the "peakFilter(freq.min=0.01)".
Best,
Dmitry
----------------------------------------
#defines paths
pathcontrol <- "C:/Users/Dima/Downloads/r1s1LSliceHippo201.imzML"
pathinjured <- "C:/Users/Dima/Downloads/r2s2LSliceHippo201.imzML"
#loads data
hippocontrol<- readMSIData(pathcontrol, mass.range=c(730,830), resolution=100, units="ppm", attach.only=TRUE)
hippoinjured<- readMSIData(pathinjured, mass.range=c(730,830), resolution=100, units="ppm", attach.only=TRUE)
pathcontrol <- "C:/Users/Dima/Downloads/r1s1LSliceHippo201.imzML"
pathinjured <- "C:/Users/Dima/Downloads/r2s2LSliceHippo201.imzML"
#loads data
hippocontrol<- readMSIData(pathcontrol, mass.range=c(730,830), resolution=100, units="ppm", attach.only=TRUE)
hippoinjured<- readMSIData(pathinjured, mass.range=c(730,830), resolution=100, units="ppm", attach.only=TRUE)
#pre-processing for control and injured
hippocontrol_pre <- hippocontrol %>% normalize(method="rms", delay=TRUE) %>% peakFilter(freq.min=0.01)
processingData(hippocontrol_pre)
hippocontrol_proc <- process(hippocontrol_pre,centroided=TRUE,plot=TRUE)
hippoinjured_pre <- hippoinjured %>% normalize(method="rms", delay=TRUE) %>% peakFilter(freq.min=0.01)
processingData(hippoinjured_pre)
hippoinjured_proc <- process(hippoinjured_pre,centroided=TRUE,plot=TRUE)
hippocontrol_pre <- hippocontrol %>% normalize(method="rms", delay=TRUE) %>% peakFilter(freq.min=0.01)
processingData(hippocontrol_pre)
hippocontrol_proc <- process(hippocontrol_pre,centroided=TRUE,plot=TRUE)
hippoinjured_pre <- hippoinjured %>% normalize(method="rms", delay=TRUE) %>% peakFilter(freq.min=0.01)
processingData(hippoinjured_pre)
hippoinjured_proc <- process(hippoinjured_pre,centroided=TRUE,plot=TRUE)
-----------------------------------------------------
Dmitry Leontyev
Feb 7, 2022, 4:20:18 PM2/7/22
to Cardinal MSI Help
After removing the
"peakFilter(freq.min=0.01)", I was able to get the control and injured processed files to have the same number of features and mass range, but I am still getting the same error as before when trying to combine:
On Monday, February 7, 2022 at 2:15:47 PM UTC-5 educh...@gmail.com wrote:
Gordon Luu
Feb 7, 2022, 5:02:23 PM2/7/22
to Cardinal MSI Help
Personally I use the "Export Spectra from Disk Option", which doesn't do any processing from flexImaging. I also combine using the Cardinal::combine() function, not cbind, and I do this prior to any preprocessing in Cardinal.
Dmitry Leontyev
Feb 8, 2022, 9:06:31 AM2/8/22
to Cardinal MSI Help
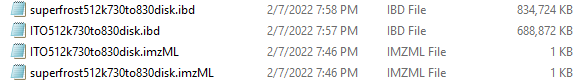
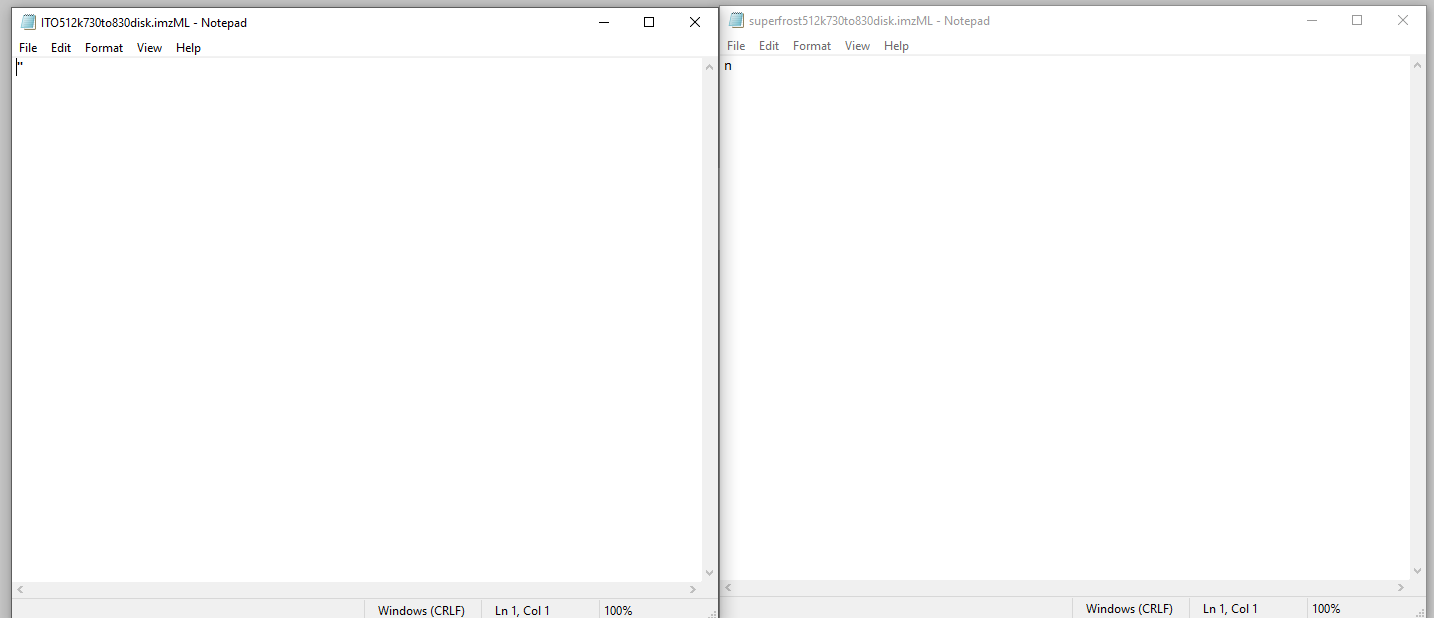
When I export data from flexImaging using the disk option, my resulting .imzML files are 1KB in size and when I open them in notepad, they just have 1 random character in them. When I export using the processed option, both my .ibd and .imzML files are 1KB in size and essentially blank. Any clue what might be going on here?

Dmitry Leontyev
Feb 8, 2022, 9:43:38 AM2/8/22
to Cardinal MSI Help
Since flexImaging is giving me some issues, I went back to try to combine data sets before processing and am getting the same error I always get when trying to use combine(). Are you familiar with this error?

--------------------------------
#defines paths
pathcontrol <- "C:/Users/Dima/Downloads/r1s1LSliceHippo201.imzML"
pathinjured <- "C:/Users/Dima/Downloads/r2s2LSliceHippo201.imzML"
#loads data
pathcontrol <- "C:/Users/Dima/Downloads/r1s1LSliceHippo201.imzML"
pathinjured <- "C:/Users/Dima/Downloads/r2s2LSliceHippo201.imzML"
#loads data
hippocontrol<- readMSIData(pathcontrol, mass.range=c(730,830), resolution=100, units="ppm", centroided=TRUE, attach.only=TRUE)
hippoinjured<- readMSIData(pathinjured, mass.range=c(730,830), resolution=100, units="ppm", centroided=TRUE, attach.only=TRUE)
combinedRun <- combine(hippocontrol,hippoinjured,centroided=TRUE)
hippoinjured<- readMSIData(pathinjured, mass.range=c(730,830), resolution=100, units="ppm", centroided=TRUE, attach.only=TRUE)
combinedRun <- combine(hippocontrol,hippoinjured,centroided=TRUE)
------------------------------------------------
On Monday, February 7, 2022 at 5:02:23 PM UTC-5 go...@ucsc.edu wrote:
Dmitry Leontyev
Feb 8, 2022, 4:13:43 PM2/8/22
to Cardinal MSI Help
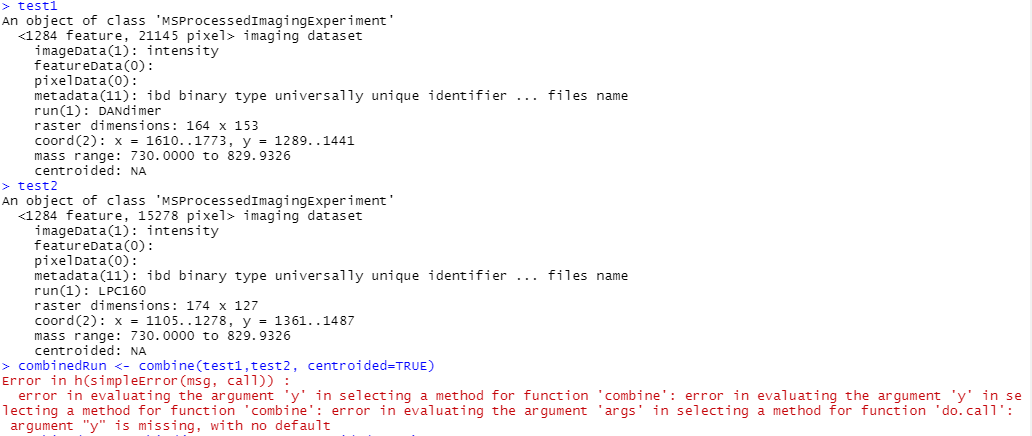
I was able to export some files from flexImaging and the z-coordinate issue went away. But combine() still gives the same error (code and output below). Am I calling combine() improperly?
---------Code---------------
#defines paths
pathtest1 <- "C:/Users/Dima/Downloads/DANdimer.imzML"
pathtest2 <- "C:/Users/Dima/Downloads/LPC160.imzML"
#loads data
test1 <- readMSIData(pathtest1, mass.range=c(730,830), resolution=100, units="ppm", attach.only=TRUE)
test2 <- readMSIData(pathtest2, mass.range=c(730,830), resolution=100, units="ppm", attach.only=TRUE)
combinedRun <- combine(test1,test2, centroided=TRUE)
pathtest1 <- "C:/Users/Dima/Downloads/DANdimer.imzML"
pathtest2 <- "C:/Users/Dima/Downloads/LPC160.imzML"
#loads data
test1 <- readMSIData(pathtest1, mass.range=c(730,830), resolution=100, units="ppm", attach.only=TRUE)
test2 <- readMSIData(pathtest2, mass.range=c(730,830), resolution=100, units="ppm", attach.only=TRUE)
combinedRun <- combine(test1,test2, centroided=TRUE)
--------------------------------------------
Gordon Luu
Feb 8, 2022, 4:17:16 PM2/8/22
to Cardinal MSI Help
The should be:
combinedRun <- Cardinal::combine(test1,test2, centroided=TRUE)
I believe combine() defaults to some base R function.
educh...@gmail.com
Feb 8, 2022, 4:20:52 PM2/8/22
to Cardinal MSI Help
Yes, you need to be specific when using combine function as Cardinal::combine. There is a default function in R that could give errors.
By the way, I’m using mzbin option to get the same mz range for all pixels in different tissues. Then I use combine to get all them together and generate more analysis or dataframes/csv…
Dmitry Leontyev
Feb 8, 2022, 4:23:43 PM2/8/22
to Cardinal MSI Help
I am still getting the same error. If you inspect the output of test1 and test2 (in the previous message) is there anything that stands out as unusual that would interfere with combining? Like the pixel data is empty.

Dmitry Leontyev
Feb 9, 2022, 9:58:07 AM2/9/22
to Cardinal MSI Help
Update: I was able to find the source of the combine() issue. This is because centroided was set to NA after reading in my imzML files. If after loading in files I called "centroided(file_name) <- TRUE" it would set centroided to TRUE. This would then allow me to use combine() as intended (also requires deleting centroided=TRUE from combine()). If imaging with an FTICR and exporting as the default centroided options, this should be kept in mind.

Fixed code:
Reply all
Reply to author
Forward
0 new messages