mzAlign vs PeakAlign
227 görüntüleme
İlk okunmamış mesaja atla

Ethan Yang
14 Oca 2021 20:49:0614.01.2021
alıcı Cardinal MSI Help
Hi All,
I am having some difficulties figuring out the difference between mzAlign and PeakAlign. I have always used PeakAlign after PeakPick as suggested in the Workflow, so where/when is it more appropriate to use mzAlign?
Any input/guidance would be highly appreciated!
Sincerely,
Ethan Yang

foellm...@gmail.com
18 Oca 2021 16:09:2118.01.2021
alıcı Cardinal MSI Help
Hi Ethan,
mzAlign aligns spectra in profile mode to reduce mass shifts between them and should therefore be applied before peak picking. PeakAlign is required after PeakPicking but might not change much if mzAlign was already applied. For my low mass resolution peptide imaging data I perform the three steps directly in this order (with more steps before and afterwards though): mzAlign - PeakPicking - PeakAlign
HTH
Melanie
Ethan Yang
18 Oca 2021 16:49:5418.01.2021
alıcı Cardinal MSI Help
Hi Melanie,
Thanks for sharing your workflow. It makes much more sense to me now, and I would love to make use of this function as a post acquisition internal calibration step. However, after looking through the thread and other documentation, it seems that this function is currently still only able to take in reference spectrum, so we will have to create a synthetic spectrum for this function. Is this your approach as well?
Sincerely,
foellm...@gmail.com
21 Oca 2021 08:43:3721.01.2021
alıcı Cardinal MSI Help
True there is also re-calibration to bring the peaks into a correct position on the m/z axis. In the latest Cardinal version (2.8) it is possible to just provide the known m/z values for re-calibration instead of a synthetic spectrum.
So I first align the shits between the spectra and then bring the peaks to the right m/z position in a second step. Unfortunately, currently these provided m/z values will stay a sort of a "reference", therefore after peak picking it's important to create an average spectrum which is then used as reference during peakAlignment.
Here is some exemplary code:
## alignment
TMAs_mzaligned <- mzAlign(TMAs_s_b, tolerance = 200, units = "ppm")
TMAs_mzaligned <- process(TMAs_mzaligned)
## mz recalibration
calibrant_mz <- c(805.42, 1296.69, 1347.72, 1570.68)
TMAs_recalibrated <- mzAlign(TMAs_mzaligned, ref=calibrant_mz, tolerance=200, units="ppm")
TMAs_recalibrated <- process(TMAs_recalibrated)
## peak picking, alignment, filtering
TMAs_picked <- peakPick(TMAs_recalibrated, method = "simple", SNR=5, window = 10, blocks = 500)
TMAs_picked <- process(TMAs_picked)
mean_MSI <- summarizeFeatures(TMAs_picked, FUN="mean")
fData(TMAs_picked)$mean_spectrum <- as.numeric(spectra(mean_MSI))
TMAs_pp <- peakAlign(TMAs_picked, ref="mean_spectrum", tolerance = 200, units = "ppm")
TMAs_pp <- peakFilter(TMAs_pp, freq.min = 0.01)
TMAs_pp <- process(TMAs_pp)
Ethan Yang
21 Oca 2021 18:51:3021.01.2021
alıcı Cardinal MSI Help
Hi Melanie,
Thanks for the example code! I noticed that you have provided 4 peaks. Is there a recommended amount? What kind of algorithm is used in this process? Is it something similar to the quadratic/cubic realignment when we do initial calibration before data acquisition?
Sincerely,

educh...@gmail.com
26 Oca 2021 04:29:5326.01.2021
alıcı Cardinal MSI Help
Hi Ethan,
the most reference peaks the better. You need to cover the entire range with them, ie, if you have a scan between 700-2000, distribute 4-5 peaks more or less uniformly over this range (750, 1100, 1500, 1900).
I've a question to both of you, Melanie and Ethan: when i perform my recalibration with Melanie code, RStudio session just crashed and doesn't work. Did you tried it? I've Cardinal2.6.0 installed and 256GB RAM available
This is my code:
#refpeaks
calibrant_mz <- c(740.40, 842.51, 1046.54, 1570.68)
#Alignm
tissue_mzAlign<-mzAlign(tissue_pre, tolerance = 2000, units = "ppm")
tissue _mzAlign<-process(tissue _mzAlign) ######HERE RSTUDIO CRASH#####
#Recal
tissue _recal<-mzAlign(tissue _mzAlign, ref=calibrant_mz,
calibrant_mz <- c(740.40, 842.51, 1046.54, 1570.68)
#Alignm
tissue_mzAlign<-mzAlign(tissue_pre, tolerance = 2000, units = "ppm")
tissue _mzAlign<-process(tissue _mzAlign) ######HERE RSTUDIO CRASH#####
#Recal
tissue _recal<-mzAlign(tissue _mzAlign, ref=calibrant_mz,
tolerance = 200, units = "ppm")
tissue
_recal<- process(tissue
_recal)
#picking
tissue _picked <- peakPick(tissue _recal, method = "simple", SNR=5, window = 10, blocks = 500)
tissue_picked <- process(tissue _picked)
#peakAlign
mean_MSI <- summarizeFeatures(tissue _picked, FUN="mean")
fData(tissue _picked)$mean_spectrum <- as.numeric(spectra(mean_MSI))
#picking
tissue _picked <- peakPick(tissue _recal, method = "simple", SNR=5, window = 10, blocks = 500)
tissue_picked <- process(tissue _picked)
#peakAlign
mean_MSI <- summarizeFeatures(tissue _picked, FUN="mean")
fData(tissue _picked)$mean_spectrum <- as.numeric(spectra(mean_MSI))
Best regards!

kbemis
26 Oca 2021 10:44:4926.01.2021
alıcı Cardinal MSI Help
As Edu says, the more reference peaks, the better. There is no recommended amount. One would have to perform an experiment with known calibrated data and references and compare the results.
This is an experimental, unpublished method, although the implementation is fairly standard. It uses local regression to estimate the mass shifts, and then cubic spline interpolation to perform the spectral shifts.
-Kylie
Melanie Föll
26 Oca 2021 14:44:5826.01.2021
alıcı Cardinal MSI Help
Hi Edu,
my mzAlign code will only work in Cardinal v2.8. For v2.6 you need to generate a reference spectrum of your calibrants as described in another conversation here.
Best,
Melanie

ismaelu...@gmail.com
2 Mar 2021 04:39:032.03.2021
alıcı Cardinal MSI Help
Hi Melanie,
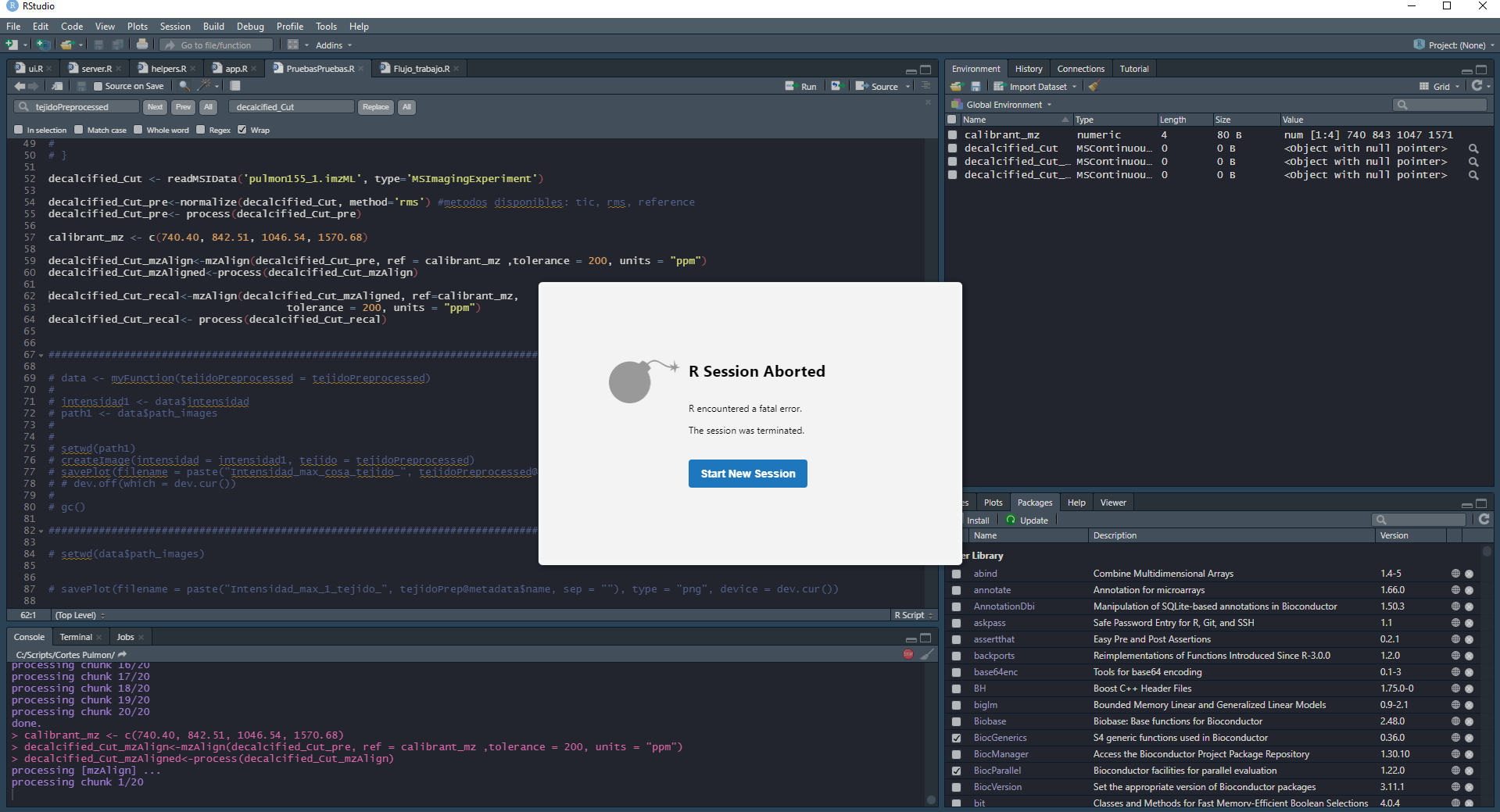
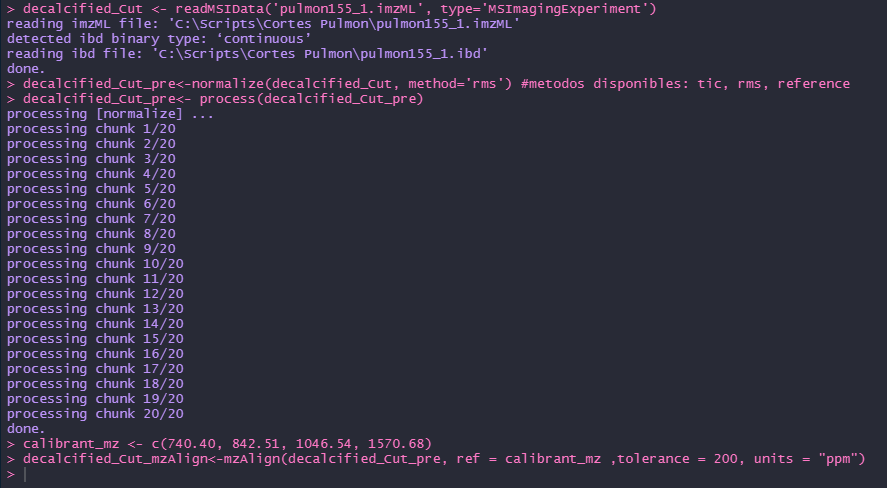
I'm working with Edu in the mzalign code but we have a problem running the code, I'm working with 2.8v of Cardinal, when we run the code (below) the R session just stopped.
I let you one pic of the error.
## CODE ##
decalcified_Cut <- readMSIData('pulmon155_1.imzML', type='MSImagingExperiment')
decalcified_Cut_pre<-normalize(decalcified_Cut, method='rms') #metodos disponibles: tic, rms, reference
decalcified_Cut_pre<- process(decalcified_Cut_pre)
calibrant_mz <- c(740.40, 842.51, 1046.54, 1570.68)
decalcified_Cut_mzAlign<-mzAlign(decalcified_Cut_pre, ref = calibrant_mz ,tolerance = 200, units = "ppm")
decalcified_Cut_mzAligned<-process(decalcified_Cut_mzAlign)
decalcified_Cut_recal<-mzAlign(decalcified_Cut_mzAligned, ref=calibrant_mz,
tolerance = 200, units = "ppm")
decalcified_Cut_recal<- process(decalcified_Cut_recal)
Thanks for your attention.
- Ismael
{kind=link}
{kind=link}
Melanie Föll
11 Mar 2021 04:03:3711.03.2021
alıcı Cardinal MSI Help
Hi Ismael,
your code looks fine to me but unfortunately the error doesn't give any information. Maybe you were running out of memory. You could crop your dataset to a few (hundred) spectra and test the pre-processing pipeline with the small dataset before you run it on the complete data.
Best,
Melanie
İleti silindi
ismaelu...@gmail.com
15 Mar 2021 03:15:0315.03.2021
alıcı Cardinal MSI Help
So much thank you Melani, that was our problem.
When we proved it my computer don't have the enought memory and Edu had a previous version of Cardinal and made conflict but we didn't know, so we thought that the problem was another thing.
Thanks once again.
Best,
Ismael
Tümünü yanıtla
Yazarı yanıtla
Yönlendir
0 yeni ileti