Bad scaffolding. What others parameters can I tweak ?
136 views
Skip to first unread message

picas...@gmail.com
Sep 12, 2021, 5:55:20 AM9/12/21
to 3D Genomics
Hi,
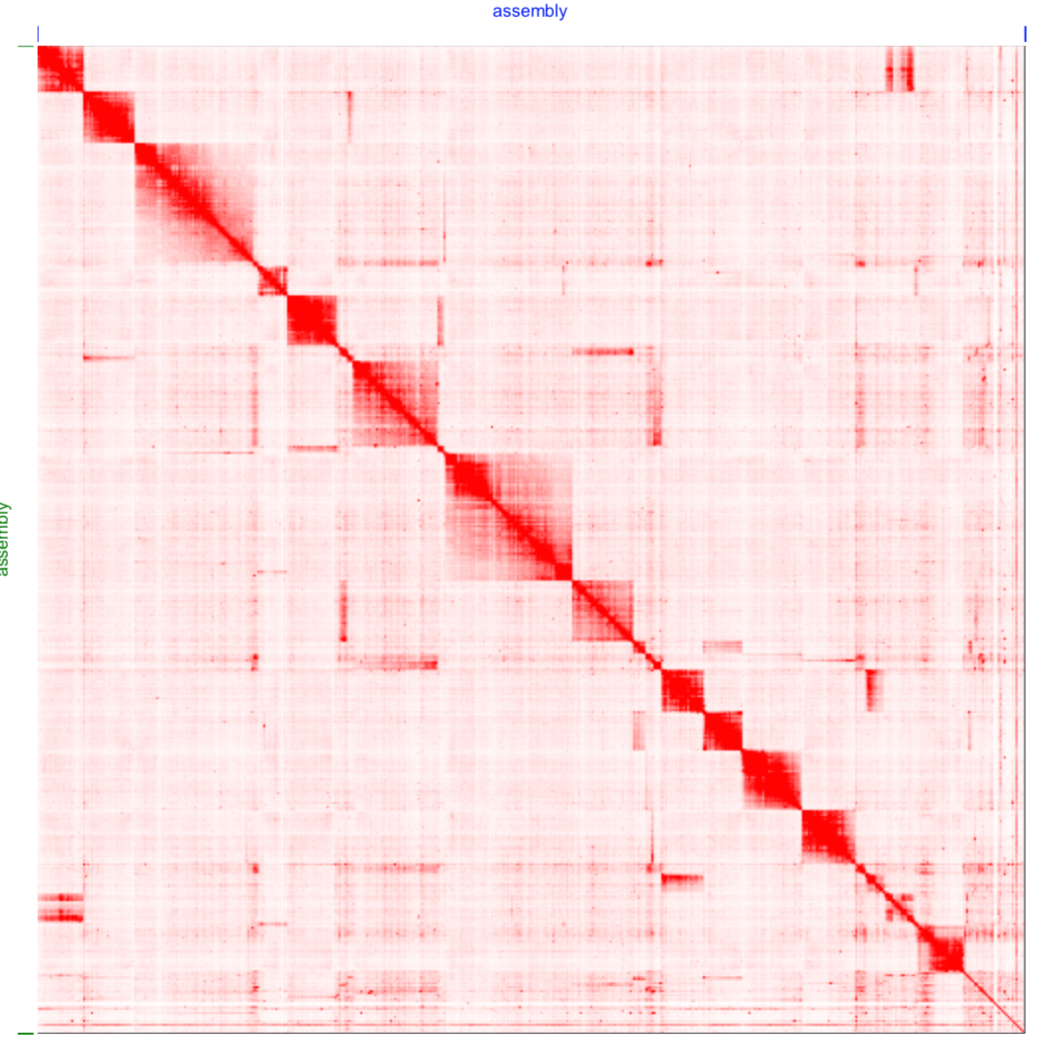
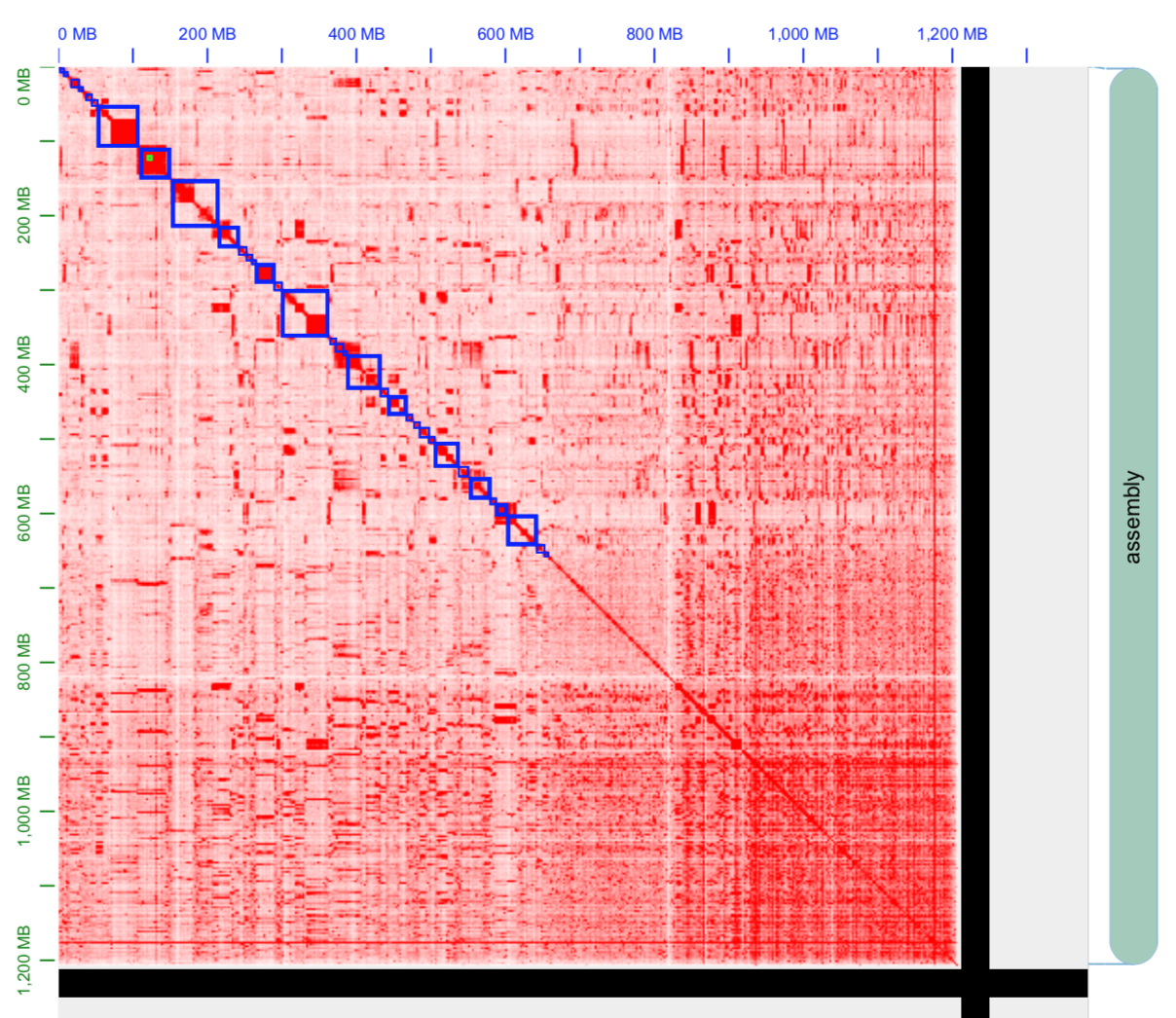
Thanks for your amazing software. I have a genome assembly at contig level which is good (high busco and haploid), but the hic scaffolding gave me vey bad result.
The default parameters are useless and I have been able to do "something" with:
--editor-repeat-coverage 10 --editor-coarse-resolution 100000 --editor-coarse-region 500000
--editor-repeat-coverage 10 --editor-coarse-resolution 100000 --editor-coarse-region 500000
Please see the default and the parameters heatmaps.
Here is the inter.txt:
Sequenced Read Pairs: 426,026,176
Normal Paired: 186,398,408 (43.75%)
Chimeric Paired: 33,452,211 (7.85%)
Chimeric Ambiguous: 66,133,845 (15.52%)
Unmapped: 140,041,712 (32.87%)
Ligation Motif Present: 0 (0.00%)
Alignable (Normal+Chimeric Paired): 219,850,619 (51.60%)
WARNING: sun.reflect.Reflection.getCallerClass is not supported. This will impact performance.
WARN [2021-09-03T17:22:07,489] [Globals.java:138] [main] Development mode is enabled
Unique Reads: 186,519,246 (43.78%)
PCR Duplicates: 32,862,575 (7.71%)
Optical Duplicates: 468,798 (0.11%)
Library Complexity Estimate: 657,152,159
Intra-fragment Reads: 0 (0.00% / 0.00%)
Below MAPQ Threshold: 85,810,163 (20.14% / 46.01%)
Hi-C Contacts: 100,709,083 (23.64% / 53.99%)
Ligation Motif Present: 0 (0.00% / 0.00%)
3' Bias (Long Range): 50% - 50%
Pair Type %(L-I-O-R): 25% - 25% - 25% - 25%
Inter-chromosomal: 51,725,911 (12.14% / 27.73%)
Intra-chromosomal: 48,983,172 (11.50% / 26.26%)
Short Range (<20Kb): 40,060,851 (9.40% / 21.48%)
Long Range (>20Kb): 8,915,404 (2.09% / 4.78%)
Normal Paired: 186,398,408 (43.75%)
Chimeric Paired: 33,452,211 (7.85%)
Chimeric Ambiguous: 66,133,845 (15.52%)
Unmapped: 140,041,712 (32.87%)
Ligation Motif Present: 0 (0.00%)
Alignable (Normal+Chimeric Paired): 219,850,619 (51.60%)
WARNING: sun.reflect.Reflection.getCallerClass is not supported. This will impact performance.
WARN [2021-09-03T17:22:07,489] [Globals.java:138] [main] Development mode is enabled
Unique Reads: 186,519,246 (43.78%)
PCR Duplicates: 32,862,575 (7.71%)
Optical Duplicates: 468,798 (0.11%)
Library Complexity Estimate: 657,152,159
Intra-fragment Reads: 0 (0.00% / 0.00%)
Below MAPQ Threshold: 85,810,163 (20.14% / 46.01%)
Hi-C Contacts: 100,709,083 (23.64% / 53.99%)
Ligation Motif Present: 0 (0.00% / 0.00%)
3' Bias (Long Range): 50% - 50%
Pair Type %(L-I-O-R): 25% - 25% - 25% - 25%
Inter-chromosomal: 51,725,911 (12.14% / 27.73%)
Intra-chromosomal: 48,983,172 (11.50% / 26.26%)
Short Range (<20Kb): 40,060,851 (9.40% / 21.48%)
Long Range (>20Kb): 8,915,404 (2.09% / 4.78%)
Do you have an explanation about the bad result and do you have other recommendation for parameters ?
Thanks a lot

{kind=link}
{kind=link}
Olga Dudchenko
Sep 14, 2021, 3:39:10 AM9/14/21
to 3D Genomics
Hello,
Standard recommendation for this is to examine .0.hic and the associated tracks bed and wig tracks. This will tell you if 1) you have decent HI-C signal (you do as far as I can tell) 2) if you data's adequate near the diagonal and what gets annotated as far as misjoin algorithm is concerned (standard issue: data is too sparse near the diagonal and the signal is not saturated. Solution: do misjoin detection at lower resolution as compared to standard) 3) if you data has large coverage biases and what gets annotated as far as repeat annotation algorithm is concerned (standard issue: average coverage is off 1x due to large repetitive content, aneuploidy, alt haplotypes etc. Standard solution: adjust editor repeat coverage).
Good luck,
Olga
Reply all
Reply to author
Forward
0 new messages