The contact map is only half size of my assembly
97 views
Skip to first unread message

YT Chen
Jul 5, 2021, 3:37:28 AM7/5/21
to 3D Genomics
Hi,
Thank you very much for developing such a nice tool for the community.
I am trying to use 3d-DNA pipeline to assemble a plant genome which is known to be 2.6 gb large. I got a 2.7 gb assembly from ONT long reads and the contig N50 is around 660 kb. Then I aligned Hi-C reads to the assembly using juicer. Below is the statistics of the hi-c alignment.
Sequenced Read Pairs: 539,516,521
Normal Paired: 213,369,608 (39.55%)
Chimeric Paired: 100,242,870 (18.58%)
Chimeric Ambiguous: 218,009,336 (40.41%)
Unmapped: 7,894,707 (1.46%)
Ligation Motif Present: 373,605,690 (69.25%)
Alignable (Normal+Chimeric Paired): 313,612,478 (58.13%)
Unique Reads: 290,267,405 (53.80%)
PCR Duplicates: 23,239,355 (4.31%)
Optical Duplicates: 105,565 (0.02%)
Library Complexity Estimate: 2,008,809,080
Intra-fragment Reads: 25,254,264 (4.68% / 8.70%)
Below MAPQ Threshold: 190,171,454 (35.25% / 65.52%)
Hi-C Contacts: 74,841,687 (13.87% / 25.78%)
Ligation Motif Present: 57,935,480 (10.74% / 19.96%)
3' Bias (Long Range): 65% - 35%
Pair Type %(L-I-O-R): 24% - 26% - 25% - 24%
Inter-chromosomal: 37,878,703 (7.02% / 13.05%)
Intra-chromosomal: 36,962,984 (6.85% / 12.73%)
Short Range (<20Kb): 33,783,522 (6.26% / 11.64%)
Long Range (>20Kb): 3,178,444 (0.59% / 1.10%)
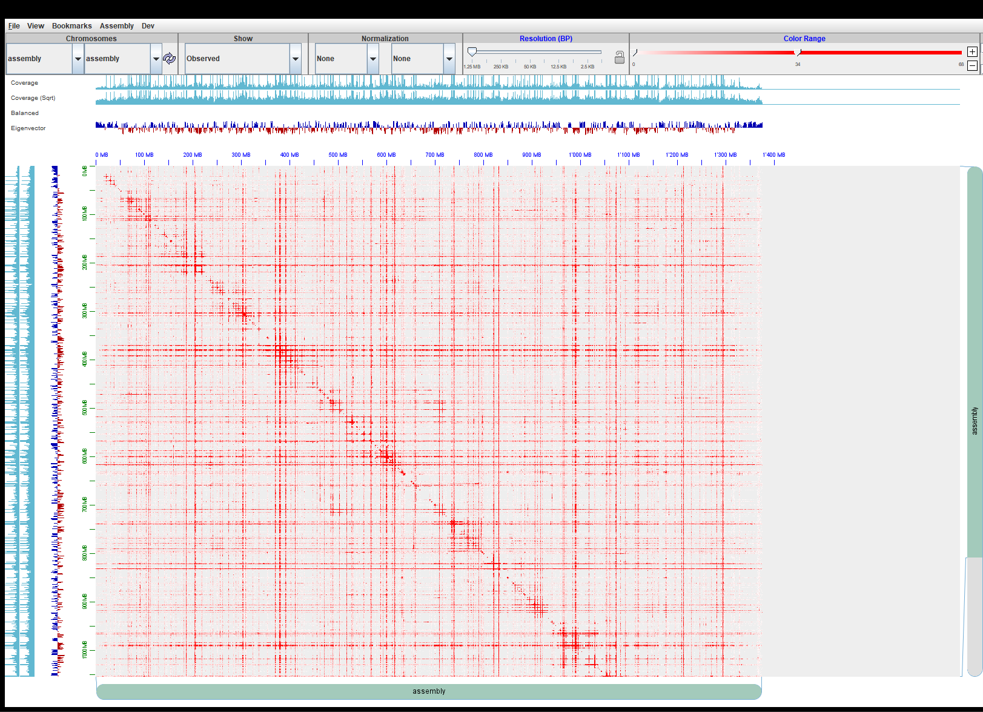
After the alignment, I tried 3d-DNA pipeline to scaffold the assembly with the Hi-C data.
I used --editor-repeat-coverage 5, --editor-coarse-resolution 100000, --splitter-coarse-stringency 30 and I got the results. Please see attached 0.hic map.
As you can see the map is only 1400 Mb but my assembly is 2700 Mb. What could be wrong? And do you have any more suggestions on which parameter I could tune as now I don't see any chromosome-scaffold patterns. Thank you very much.
Best wishes,
Yutang

{kind=link}
Olga Dudchenko
Jul 6, 2021, 1:55:00 AM7/6/21
to 3D Genomics
Hi Yutang,
The size of the map is not a concern: there is a rescaling that 3d-dna internally does to meet some internal igv requirements that's explained elsewhere. Once you load the assembly file this will be automatically adjusted. The issue here is that this map looks like it does not have any Hi-C signal in that your experiment has probably failed. What is this exactly? is this .0.hic?
Olga
YT Chen
Jul 7, 2021, 3:40:59 AM7/7/21
to 3D Genomics
Hi Olga,
Thank you very much for your explanation. The picture is 0.hic.
The Hi-C data were passed to me by a colleague who has left our group recently, so I am not so sure about the data quality.
Do you think could it be that I gave the wrong restriction enzyme to juicer? I used DpnII as my colleague told me.
I will contact my colleague to ask more details of the data.
Best wishes,
Yutang
Reply all
Reply to author
Forward
0 new messages