too many fragments despite improved contiguity of the assembly

F. Gözde Çilingir
Hello,
Thank you very much for these super useful tools. I have been working on the chromosome-level assembly of my study species’ reference genome for a while and I learned a lot by reading the papers and manuals of both Juicer and the 3D-DNA pipeline.
My focal species is a tortoise, which has an estimated genome size and heterozygosity of ~2.4Gb and 0.73%, respectively. I have a draft genome, assembled with ~20x HIFi reads which yielded a contig N50 of 62Mb, longest contig of 210Mb, and the total # of contigs were 422. When compared to the already available reference genomes that are phylogenetically close to my species, these results were so promising and I carried on with the chromosome level assembly with ~72x HiC data. Both Juicer and 3D-DNA pipeline were run with default parameters.
The inter.txt file summarised the alignment of HiC reads as follows:
Sequenced Read Pairs: 650,786,203
Normal Paired: 424,513,741 (65.23%)
Chimeric Paired: 163,140,498 (25.07%)
Chimeric Ambiguous: 46,205,161 (7.10%)
Unmapped: 16,926,803 (2.60%)
Ligation Motif Present: 0 (0.00%)
Alignable (Normal+Chimeric Paired): 587,654,239 (90.30%)
Unique Reads: 434,031,662 (66.69%)
PCR Duplicates: 142,765,112 (21.94%)
Optical Duplicates: 10,857,465 (1.67%)
Library Complexity Estimate: 963,725,497
Intra-fragment Reads: 0 (0.00% / 0.00%)
Below MAPQ Threshold: 55,723,144 (8.56% / 12.84%)
Hi-C Contacts: 378,308,518 (58.13% / 87.16%)
Ligation Motif Present: 0 (0.00% / 0.00%)
3' Bias (Long Range): 50% - 50%
Pair Type %(L-I-O-R): 25% - 25% - 25% - 25%
Inter-chromosomal: 143,029,714 (21.98% / 32.95%)
Intra-chromosomal: 235,278,804 (36.15% / 54.21%)
Short Range (<20Kb): 91,080,656 (14.00% / 20.98%)
Long Range (>20Kb): 143,956,167 (22.12% / 33.17%)
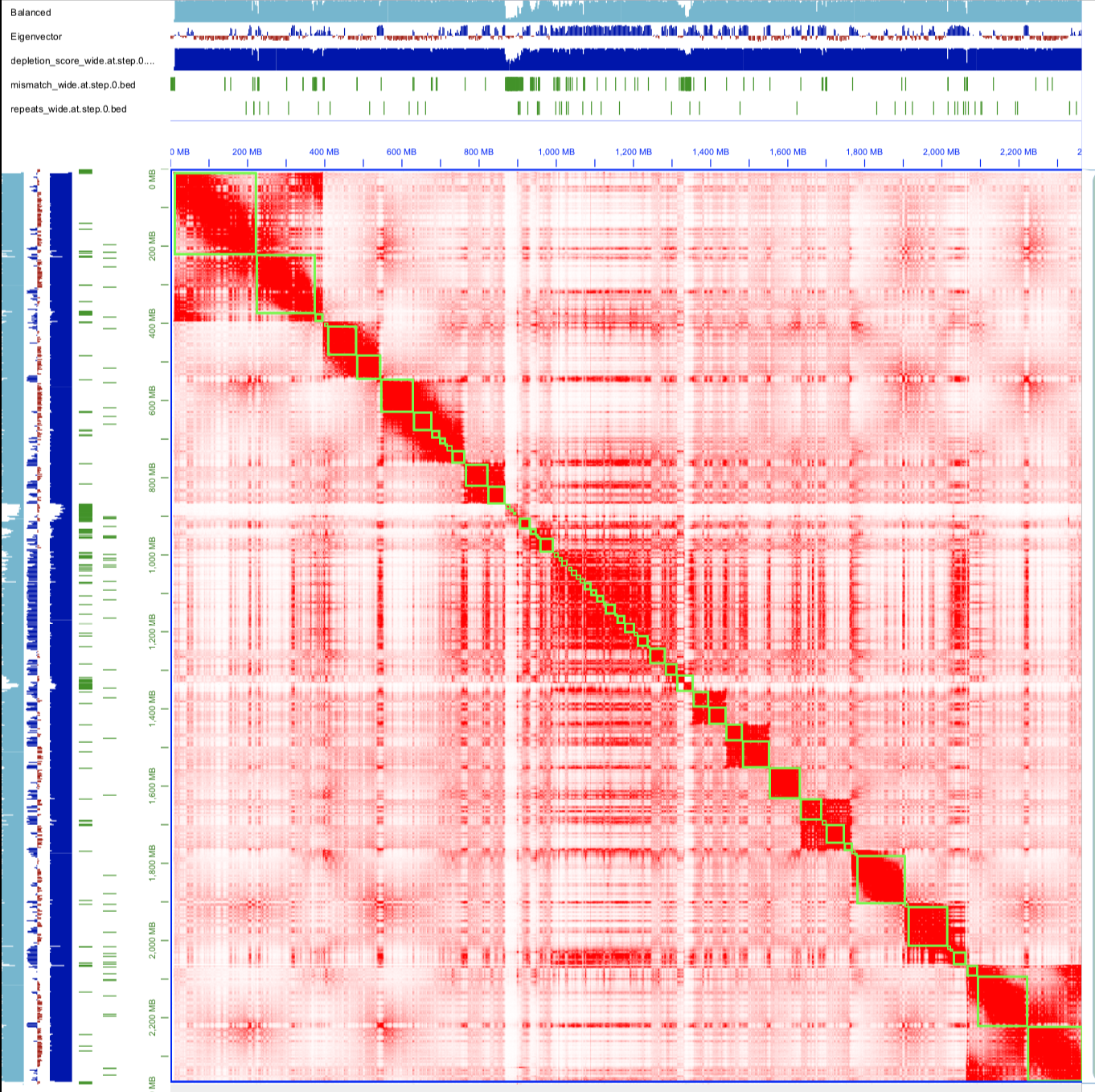
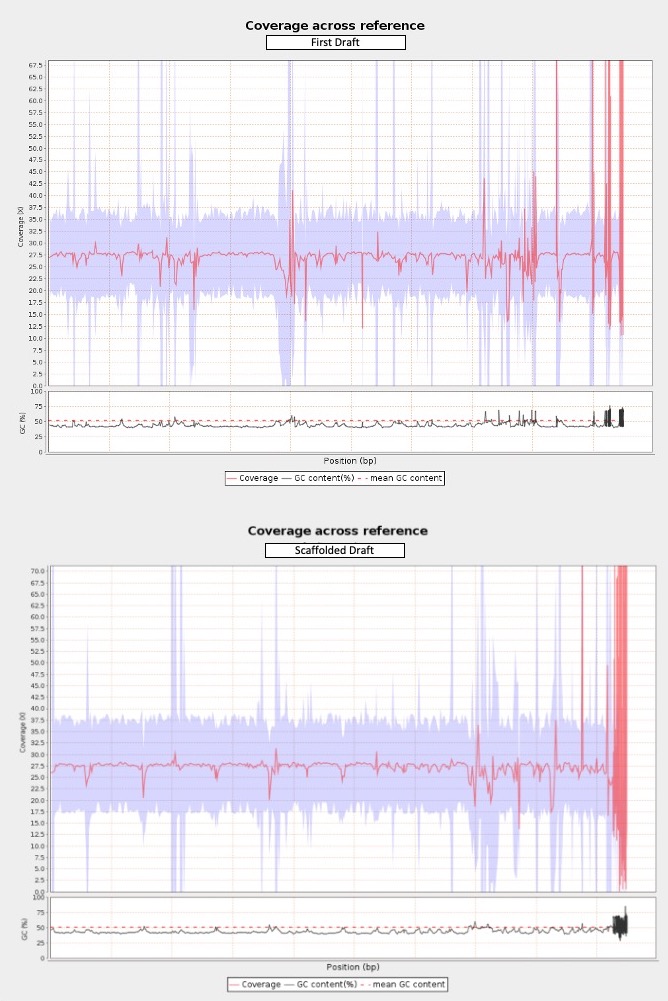
Additionally, I attached 0.hic of the 3D-DNA run coupled with additional information on coverage, depletion score and more. The total number of chromosomes was in line with my expectations and the structure of the heatmap of the green sea turtle I downloaded from DNA Zoo (also thanks for the database!). Therefore, I ran the whole pipeline. The final fasta of this run yielded a scaffold N50 of 148 Mb, longest scaffold of 383Mb which are great, however, the total number of scaffolds was 719 as opposed to the first draft’s 422. I have ~27x Illumina reads to map on the assemblies for assessment purposes, it seems like the debris from misjoins were added at the end of the assembly (please see attached), am I correct? If so, how can I decide if these are true misjoins or false positives? This tortoise species has too many minichromosomes and most of the misjoin flags seem to be from those regions. Could this be anything related to the repeat regions? Overall, why do you think there are too many fragments in my final assembly and how can I handle this situation?
Thanks a lot in advance for helping me!
Gözde

Olga Dudchenko

{kind=link}
{kind=link}
Sefa AYTEN
"In such cases it is often useful to pay attention to the percentage of reads containing ligation junctions in the raw fastq files of the Hi-C library. The exact number depends on the restriction enzyme, size selection protocols and the sequencing read length, but typically amounts to 20-40% for a reasonably good in situ Hi-C library. Low numbers may indicate poor Hi-C data quality."
I am new to work on these pipelines, but I am seeing Ligation Motif Present: 0 (0.00%) in alignment summary. Are you sure the quality of HiC reads good?
Best wishes,
Sefa