Salmon with single-end read: setting mean and std of insert length if distribution is not normal
55 views
Skip to first unread message

dmr210
Jul 19, 2017, 10:05:08 AM7/19/17
to Sailfish Users Group
Hi,
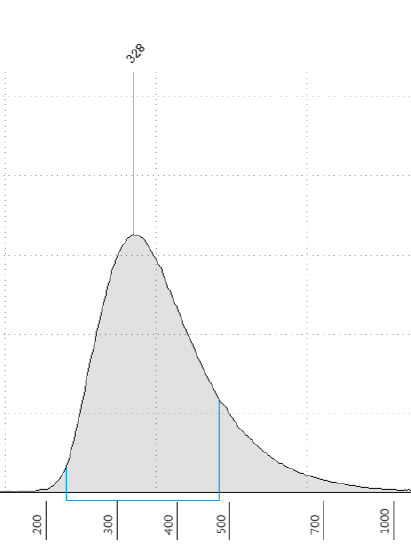
I am using salmon with single-end RNAseq for the first time. I obtained the distribution of fragment lengths for my libraries in order to provide salmon with the mean and std of the insert length, however the distribution is not normal but rather skewed towards higher fragment lengths (see screenshot of one of the libraries attached).
I have two questions:
1- Will this be an issue for the quantification by Salmon? And should I revert to alignment based quantification for this particular case to avoid issues?
2- If it is ok to use with the pseudo-alignment method, which value should I use for mean and std? Should I ignore the skewness and give that value that corresponds to the maximum density or should I give the actual mean of my distribution? And how do I deal with the fact that the std will not account for the skewness at all?
Thanks very much
Delphine
{kind=link}
Reply all
Reply to author
Forward
0 new messages