Deletion treated as Intron
65 views
Skip to first unread message

naresh prodduturi
Jun 2, 2017, 3:10:07 PM6/2/17
to rna-star
For some reason, star is aligning RNA-seq reads over the known deletion region (24 bp) as an intron instead of deletion. This deletion is confirmed in the DNA.
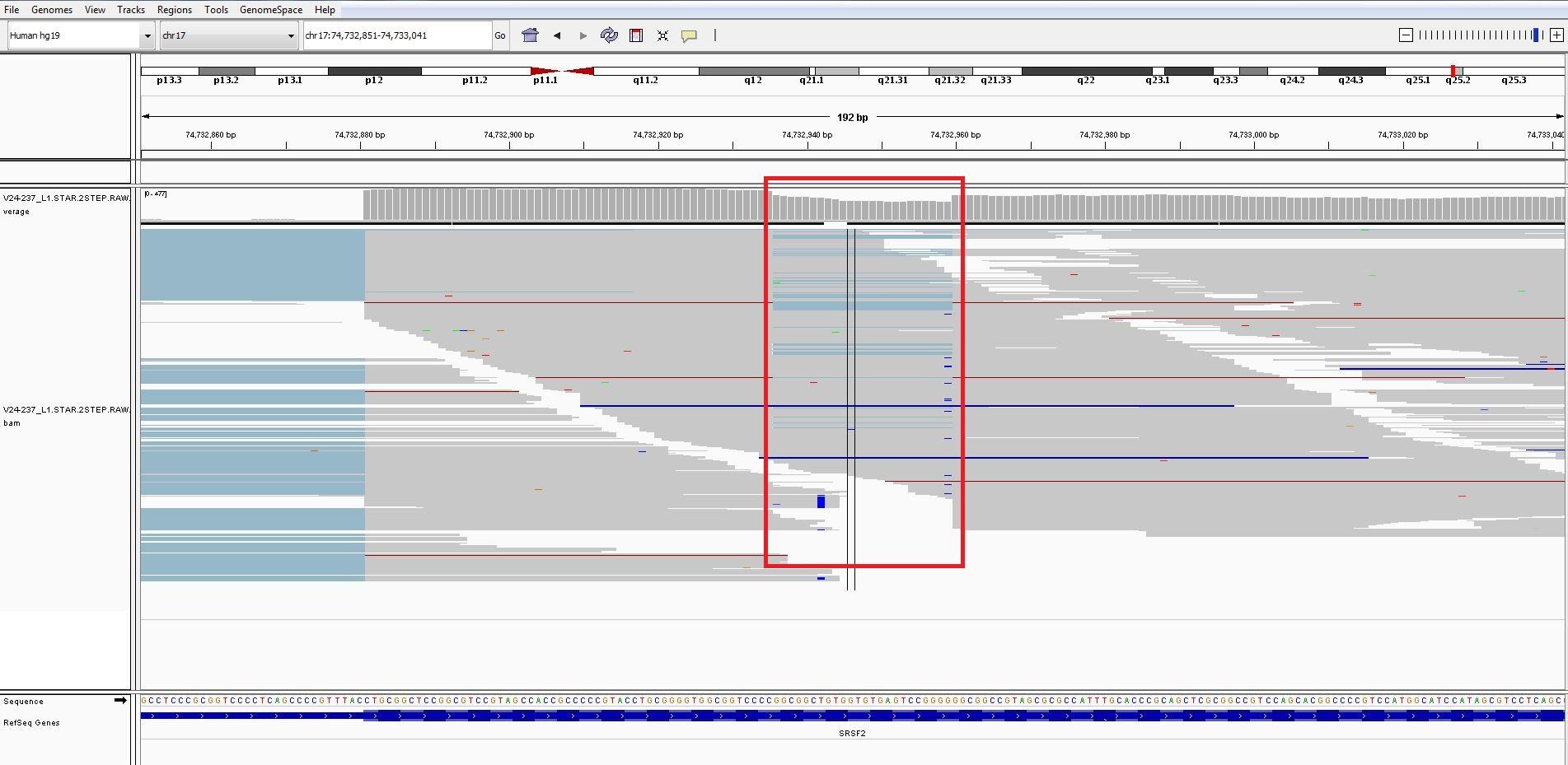
Attached is the IGV image.
i tried increasing the intron size option to 50 "--alignIntronMin 50" but still reads are aligned as intron instead of deletion
alignIntronMin 21
minimum intron size: genomic gap is considered intron if its length>=alignIntronMin, otherwise it is considered Deletion
Naresh

{kind=link}
Alexander Dobin
Jun 2, 2017, 3:59:30 PM6/2/17
to rna-star
Hi Naresh,
--alignIntronMin 50 should have resolved it. Please check the CIGAR string in these reads - if ii has 24N and not 24D, please send me a few reads for checking.
Cheers
Alex
Alexander Dobin
Jun 8, 2017, 4:41:36 PM6/8/17
to rna-star
Hi Naresh,
I mapped the reads that contain 24N with --alignIntronMin 50 and I cannot reproduce this problem.
Is it possible that this junction is annotated in your GTF file?
Please try to map a few of these reads with --outSAMattributes NH HI AS nM jM jI, this will tell us the annotation status of these junctions.
Cheers
Alex
naresh prodduturi
Jun 9, 2017, 5:57:05 PM6/9/17
to rna-star
Here is the region chr17:74,732,798-74,733,094
I used the 2 step alignment.Finding the junctions and then the actual alignment step.Can you rerun using the 2 step star alignment.
What version of STAR you are using?
It was showing as a intron in the IGV but when i checked the SJ.out.tab, i did not find that junction
But as you mentioned, i tried running the 1 step star alignment with --outSAMattributes NH HI AS nM jM jI and --alignIntronMin 50. Now i cannot see the intron but it is missing the deletion in that region, Also the coverage is dropping in that region. Attached is the bam file.
Regards
Naresh
Alexander Dobin
Jun 12, 2017, 3:03:38 PM6/12/17
to rna-star
Hi Naresh,
if I run with --twopassMode Basic, it still does not output the 24N introns.
Did you generate the genome for the 2nd pass manually?
If so, which parameters did you use to run the 1st pass? You have to specify --alignIntronMin 50 for the 1st pass run as well.
Cheers
Alex
naresh prodduturi
Jun 13, 2017, 2:48:53 PM6/13/17
to rna-star
Thank you Alex. When i used --alignIntronMin 50 in the 1st pass, there are no more reads with 24N.
But there is no deletion in that region. (clearly there should be a deletion, i confirmed it with a GSNAP aligner and also in the DNA Bam file)
Alexander Dobin
Jun 13, 2017, 5:12:13 PM6/13/17
to rna-star
Hi Naresh,
the indels have per-base penalty (-2 by default), i.e. the penalty grows linearly with the deletion length, and for 24b deletion is too big.
If you want to consider such large deletions, you need to change to -1 (or even 0) zero with --scoreDelBase -1. This results in:
1 0 chr17 76736799 255 1S55M24D45M * 0 0 GCTGCGGCTCCGGCGTCCGTAGCCACCGCCCCCGTACCTGCGGGGTGGCGGTCCCCGGCGGCCGTAGCGCGCCATTTGCACCCGCAGCTCGCGGCCGTCCA * NH:i:1 HI:i:1 AS:i:96 nM:i:0
You may also want to change --scoreInsBase the same way. There are also --scoreInsOpen and --scoreDelOpen (=-2 by default) that add constant penalty for deletion of any length.
Cheers
Alex
naresh prodduturi
Jun 14, 2017, 11:08:17 AM6/14/17
to rna-star
How this parameter change will affect the overall alignment (expression, fusion , snv?)
Alexander Dobin
Jun 15, 2017, 11:05:39 AM6/15/17
to rna-star
Hi Naresh,
it will allow more large indels to be detected - i.e. increase sensitivity - at the cost of the increase in false positive indels.
It may you findings at some particular loci, but I think those will be very few.
Cheers
Alex
Reply all
Reply to author
Forward
0 new messages