Too many % reads unmapped too short | 55.91%
462 views
Skip to first unread message

Sneh
May 10, 2016, 12:08:27 PM5/10/16
to rna-star
Hello,
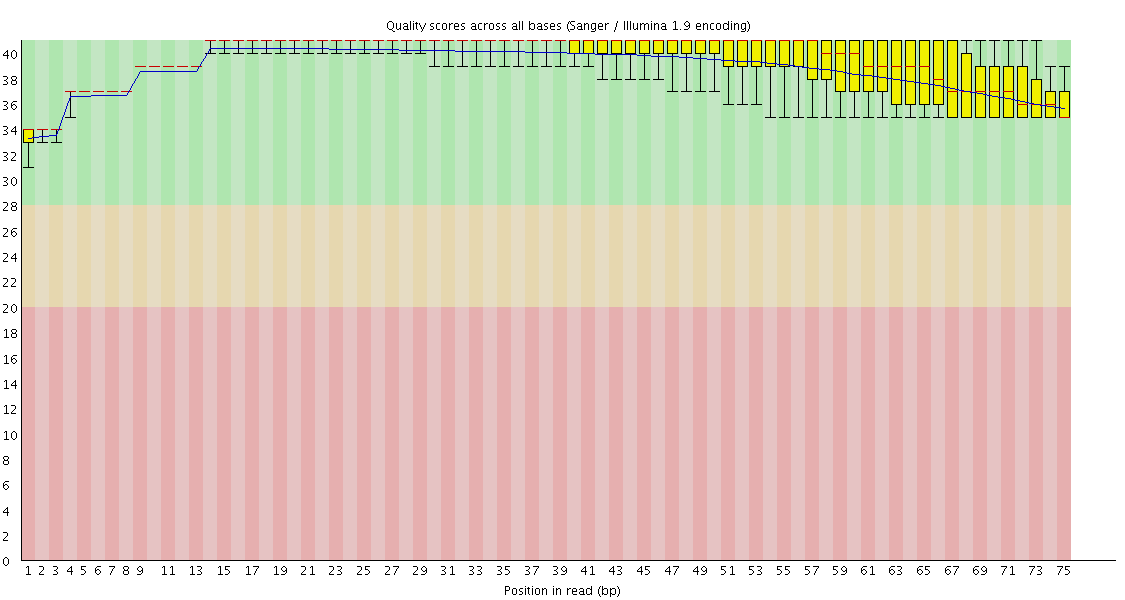
I have 75bp single end drosophila RNAseq reads. The FASTQC report the reads looks good. But when I try to align the reads to the reference dm3 genome, very little (39.16%) reads align to it. I get a very high percentage of unmapped reads because they were too short.
I tried multiple commands but nothing made much difference:
/data/administrative/Softwares/STAR-STAR_2.4.2a/bin/Linux_x86_64/STAR --genomeDir /data/administrative/Public_Databases/Reference/Drosophila/UCSC/Drosophila_melanogaster/UCSC/dm3/STAR_index/ --readFilesIn CD1.fastq
/data/administrative/Softwares/STAR-STAR_2.4.2a/bin/Linux_x86_64/STAR --genomeDir /data/administrative/Public_Databases/Reference/Drosophila/UCSC/Drosophila_melanogaster/UCSC/dm3/STAR_index/ --clip3pNbases 25 --sjdbOverhang 74 --readFilesIn CD1.fastq
Started job on | May 03 12:44:09
Started mapping on | May 03 12:49:20
Finished on | May 03 15:09:26
Mapping speed, Million of reads per hour | 4.35
Number of input reads | 10156563
Average input read length | 75
UNIQUE READS:
Uniquely mapped reads number | 3977709
Uniquely mapped reads % | 39.16%
Average mapped length | 72.43
Number of splices: Total | 104631
Number of splices: Annotated (sjdb) | 0
Number of splices: GT/AG | 100227
Number of splices: GC/AG | 848
Number of splices: AT/AC | 299
Number of splices: Non-canonical | 3257
Mismatch rate per base, % | 1.02%
Deletion rate per base | 0.02%
Deletion average length | 1.15
Insertion rate per base | 0.00%
Insertion average length | 1.87
MULTI-MAPPING READS:
Number of reads mapped to multiple loci | 461916
% of reads mapped to multiple loci | 4.55%
Number of reads mapped to too many loci | 26928
% of reads mapped to too many loci | 0.27%
UNMAPPED READS:
% of reads unmapped: too many mismatches | 0.00%
% of reads unmapped: too short | 55.91%
% of reads unmapped: other | 0.11%
I am not sure how to fix this issue. Any helpful advise is much appreciated.
Thanks
Sneh

Alexander Dobin
May 11, 2016, 1:16:04 PM5/11/16
to rna-star
Hi Sneh,
there are several reasons for high % of unmapped reads.
In your case, the sequencing quality scores are good - however, strangely, the mismatch error rate is quite high, 1% - usually we get <0.5% from Illumina these days.
Two remaining suspects:
(i) too short sequencing insert - try to trim adapters
(ii) contamination with other species, adapters, etc - try to BLAT unmapped reads
Cheers
Alex

{kind=link}
Varun Gupta
May 11, 2016, 1:22:54 PM5/11/16
to rna-star
Hi,
With the help of Alex, I have tried using parameters
--outFilterScoreMinOverLread 0 --outFilterMatchNminOverLread 0 but then specify how much you want your read to be matched let's say 65bp(out of 75bp). So use option --outFilterMatchNmin 65.
Change these options to see at what level you get your % unmapped too short resolved.
Alex can you also explain what is actual meaning of mismatch rate per base. I have never given that parameter any thought while mapping.
Thanks
Varun
With the help of Alex, I have tried using parameters
--outFilterScoreMinOverLread 0 --outFilterMatchNminOverLread 0 but then specify how much you want your read to be matched let's say 65bp(out of 75bp). So use option --outFilterMatchNmin 65.
Change these options to see at what level you get your % unmapped too short resolved.
Alex can you also explain what is actual meaning of mismatch rate per base. I have never given that parameter any thought while mapping.
Thanks
Varun
On Tuesday, May 10, 2016 at 12:08:27 PM UTC-4, Sneh wrote:
Alexander Dobin
May 16, 2016, 4:11:21 PM5/16/16
to rna-star
Hi Varun,
the mismatch rate is calculated as follows: for all reads that were mapped, the number of mismatched bases is divided by the total number of mapped bases.
Cheers
Alex
Reply all
Reply to author
Forward
0 new messages