FATAL ERROR
709 views
Skip to first unread message

Varun Gupta
Jul 29, 2013, 10:53:49 AM7/29/13
to rna-...@googlegroups.com
Hi Alex
Hope you are doing well.
So i started running star version of 2.3.1.o. But i guess I have some problems going on when it comes to memory and disk space. Please explain me what to do about it. I will tell you what i experienced.
So i have a human genome fasta files which i masked for nearly 2000 pseudogenes(masked with N). I created the genome with the following command:
/apps1/star/2.3.1o/intel/bin/STAR --runMode genomeGenerate --runThreadN 6 --genomeDir ./ --genomeFastaFiles /cork/vgupta12/GENOMES/masked_fully_with_pseudogenes_genome.fa
so my directory where my genome(suffix arrays) is stored is /cork/vgupta12/STAR/STAR_masked_fully_pseudogenes
I have a dataset of 16 tissues. I was running each of them with 1 hit,1mismatch and 1hit,3 mismatches.
This is the command i gave for mapping
/apps1/star/2.3.1o/intel/bin/STAR --runThreadN 8 --genomeDir /cork/vgupta12/STAR/STAR_masked_fully_pseudogenes --readFilesIn /cork/vgupta12/data/scroth_body_map/single_read/fastq_files/ERR030899.fastq --outSAMattributes All --outFilterMultimapNmax 1 --outFilterMismatchNmax 1
This above command i ran around 32 times(16X2), twice for each tissue. Some of them ran but some of them are either still running or failed giving me this FATAL ERROR when i check there Log.out file...
Read from SAindex: genomeSAindexNbases=14 nSAi=357913940
Genome file size: 3099328512 bytes; state: good=1 eof=0 fail=0 bad=0
SA file size: 3099328512 bytes; state: good=1 eof=0 fail=0 bad=0
nGenome=3099328512; nSAbyte=23596574257
GstrandBit=32 SA number of indices=5720381638
EXITING: fatal error trying to allocate genome arrays, exception thrown: std::bad_alloc
Possible cause 1: not enough RAM. Check if you have enough RAM 30261776657 bytes
Possible cause 2: not enough virtual memory allowed with ulimit. SOLUTION: run ulimit -v 30261776657
I know in your manual you have mentioned shared genomes thing. Is there any modification i can do in my mapping command which would make everything work fine. But i don't understand why such error occurred since i am working on the cluster.
Let me know what i can do and then i will ask my system admin accordingly.
Thanks
Regards
Varun
Hope you are doing well.
So i started running star version of 2.3.1.o. But i guess I have some problems going on when it comes to memory and disk space. Please explain me what to do about it. I will tell you what i experienced.
So i have a human genome fasta files which i masked for nearly 2000 pseudogenes(masked with N). I created the genome with the following command:
/apps1/star/2.3.1o/intel/bin/STAR --runMode genomeGenerate --runThreadN 6 --genomeDir ./ --genomeFastaFiles /cork/vgupta12/GENOMES/masked_fully_with_pseudogenes_genome.fa
so my directory where my genome(suffix arrays) is stored is /cork/vgupta12/STAR/STAR_masked_fully_pseudogenes
I have a dataset of 16 tissues. I was running each of them with 1 hit,1mismatch and 1hit,3 mismatches.
This is the command i gave for mapping
/apps1/star/2.3.1o/intel/bin/STAR --runThreadN 8 --genomeDir /cork/vgupta12/STAR/STAR_masked_fully_pseudogenes --readFilesIn /cork/vgupta12/data/scroth_body_map/single_read/fastq_files/ERR030899.fastq --outSAMattributes All --outFilterMultimapNmax 1 --outFilterMismatchNmax 1
This above command i ran around 32 times(16X2), twice for each tissue. Some of them ran but some of them are either still running or failed giving me this FATAL ERROR when i check there Log.out file...
Read from SAindex: genomeSAindexNbases=14 nSAi=357913940
Genome file size: 3099328512 bytes; state: good=1 eof=0 fail=0 bad=0
SA file size: 3099328512 bytes; state: good=1 eof=0 fail=0 bad=0
nGenome=3099328512; nSAbyte=23596574257
GstrandBit=32 SA number of indices=5720381638
EXITING: fatal error trying to allocate genome arrays, exception thrown: std::bad_alloc
Possible cause 1: not enough RAM. Check if you have enough RAM 30261776657 bytes
Possible cause 2: not enough virtual memory allowed with ulimit. SOLUTION: run ulimit -v 30261776657
I know in your manual you have mentioned shared genomes thing. Is there any modification i can do in my mapping command which would make everything work fine. But i don't understand why such error occurred since i am working on the cluster.
Let me know what i can do and then i will ask my system admin accordingly.
Thanks
Regards
Varun

Alexander Dobin
Jul 29, 2013, 4:26:27 PM7/29/13
to rna-...@googlegroups.com
Hi Varun,
this error is caused by insufficient memory allocated to the process. If you do not use shared memory, you need to allocate ~32GB of RAM for each job.
If you start 32 of these jobs simultaneously on one server, you will need 32*32GB=1024 GB, which is too much of course. On a single machine, I recommend running the jobs sequentially with number of threads equal to the humber of physical cores for each run.
If you are running these jobs on a cluster with a queuing system (like SGE), it should take care of memory allocation and not run several jobs on the same node if there is not enough memory. You need to specify the amount of memory. for example, for UGE we use:
-pe threads 16 -l m_mem_free=2G
Note that here we specify the amount of RAM per thread: 16*2G=32GB total.
Cheers
Note that here we specify the amount of RAM per thread: 16*2G=32GB total.
Cheers
Alex

chensu...@gmail.com
Aug 15, 2016, 1:02:46 PM8/15/16
to rna-star
Hi Alex,
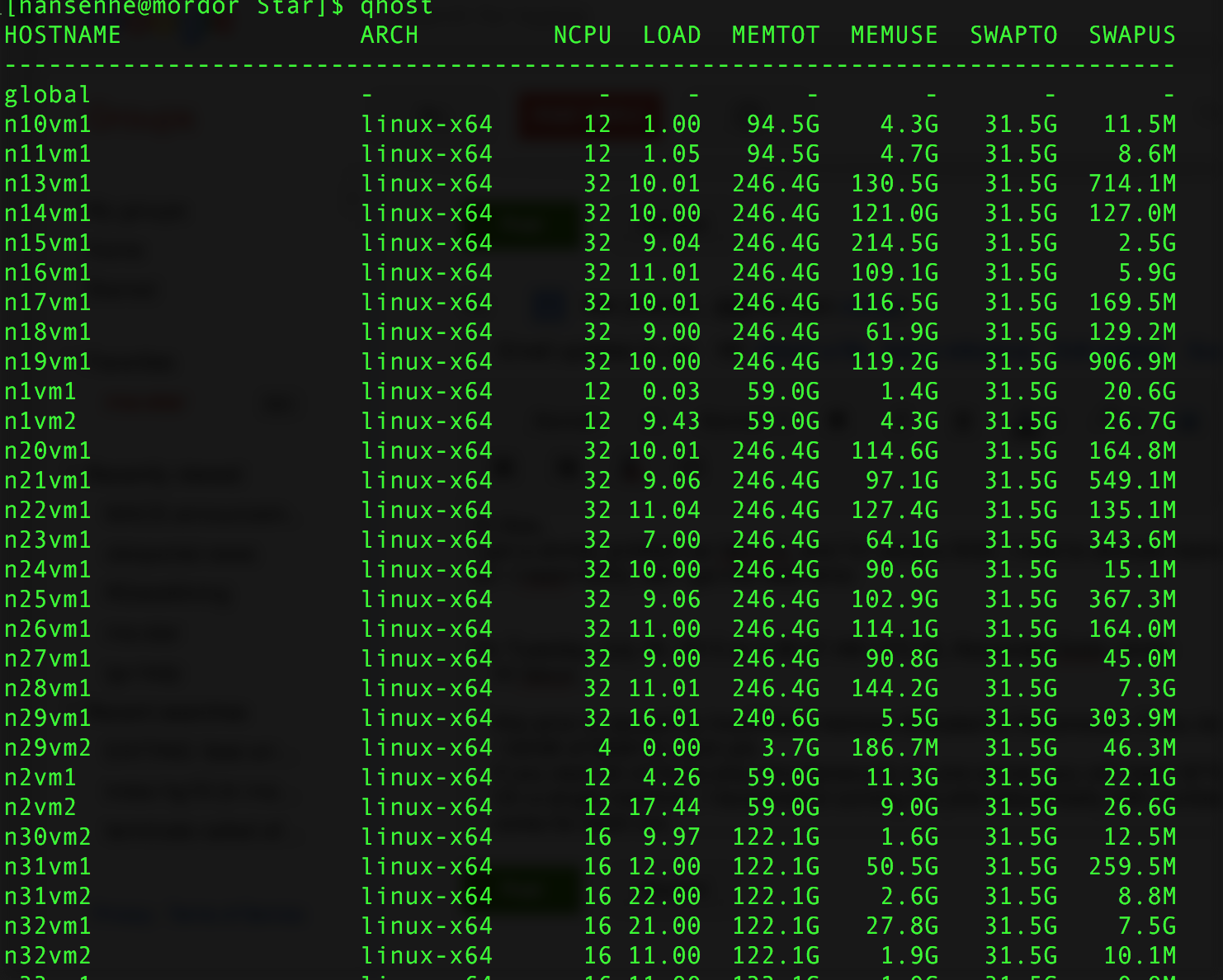
I got a similar problem as Varun's, and I'm using a SGE, I tried to allocate memory with -pe threads 16 -l m_mem_free=2G (or -l mem=32G), still got the fatal error.
The cluster usage is attached, and "Currently we do not have any policies enforced regarding restrictions on the amount of
memory, size or type of job submitted".
So can you give me a clue as what's the problem here?
Thanks for your time.
Best
Sujun
{kind=link}
Alexander Dobin
Aug 16, 2016, 4:40:09 PM8/16/16
to rna-star
Hi @chensujunotl
please send me the Log.out file of the failed run.
Cheers
Alex
Reply all
Reply to author
Forward
0 new messages