Problem with BAM QC
255 views
Skip to first unread message

Antony Vincent
Apr 27, 2015, 2:04:40 PM4/27/15
to qual...@googlegroups.com
Hello,
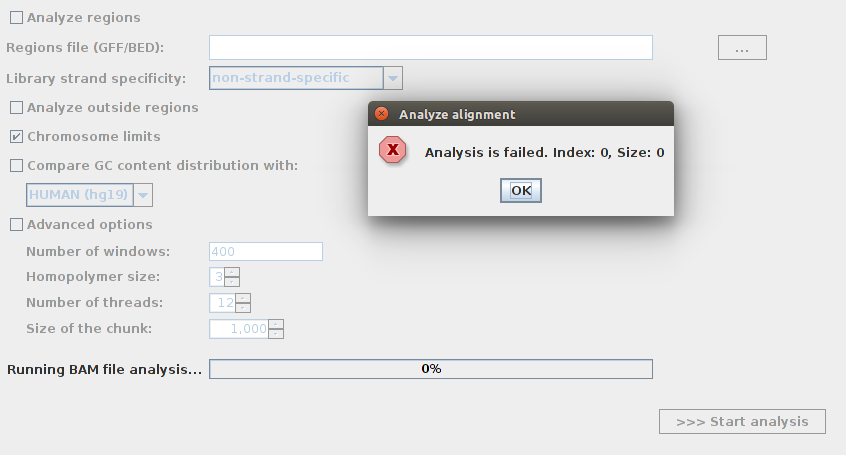
I tried to use BAM QC on BAM files to know the average coverage. For some files, when I choose to BAM file and click on " start analysis" I receive an error (please see the screenshot). Is this because there is no read mapped?
Thank you,
Antony
I tried to use BAM QC on BAM files to know the average coverage. For some files, when I choose to BAM file and click on " start analysis" I receive an error (please see the screenshot). Is this because there is no read mapped?
Thank you,
Antony

{kind=link}
Konstantin Okonechnikov
Apr 28, 2015, 8:34:12 AM4/28/15
to qual...@googlegroups.com
Hi Antony,
thanks a lot for report!
Actually, GUI might have some additional bugs issues, since command line version is used more frequently, and GUI is not used too often.
Command line has more detailed reports and better error processing.
Several questions:
1) Which Qualimap version did you use? Which operating system?
2) Did you try running analysis for the same BAM file using command line? More details here:
3) Is it possible for you to share the BAM file (or subsample from the BAM file) so I can check it carefully?
How to create subsample using samtools:
--
Konstantin
--
You received this message because you are subscribed to the Google Groups "QualiMap" group.
To unsubscribe from this group and stop receiving emails from it, send an email to qualimap+u...@googlegroups.com.
For more options, visit https://groups.google.com/d/optout.
Antony Vincent
Apr 28, 2015, 9:06:41 AM4/28/15
to qual...@googlegroups.com
Thank you for your quick answer. Here is the terminal output for the command qualimap bamqc -bam ISAS1_BWA_sorted.sam.bam -outdir qualimap_report -outformat HTML :
Java memory size is set to 1200M
Launching application...
QualiMap v.2.0
Built on 2014-08-28 17:03
Selected tool: bamqc
Available memory (Mb): 33
Max memory (Mb): 1118
Starting bam qc....
Loading sam header...
Loading locator...
Loading reference...
Number of windows: 400, effective number of windows: 304
Chunk of reads size: 1000
Number of threads: 12
Total processed windows:0
Number of reads: 1592618
Number of valid reads: 0
Number of duplicated reads: 0
Number of correct strand reads:0
Inside of regions...
Num mapped reads: 0
Num mapped first of pair: 0
Num mapped second of pair: 0
Num singletons: 0
Time taken to analyze reads: 5
Computing descriptors...
numberOfMappedBases: 0
referenceSize: 1215
numberOfSequencedBases: 0
numberOfAs: 0
Computing per chromosome statistics...
Failed to run bamqc
java.lang.IndexOutOfBoundsException: Index: 0, Size: 0
at java.util.ArrayList.rangeCheck(ArrayList.java:635)
at java.util.ArrayList.get(ArrayList.java:411)
at org.bioinfo.ngs.qc.qualimap.beans.BamStats.computeChromosomeStats(BamStats.java:1703)
at org.bioinfo.ngs.qc.qualimap.process.BamStatsAnalysis.run(BamStatsAnalysis.java:491)
at org.bioinfo.ngs.qc.qualimap.main.BamQcTool.execute(BamQcTool.java:197)
at org.bioinfo.ngs.qc.qualimap.main.NgsSmartTool.run(NgsSmartTool.java:187)
at org.bioinfo.ngs.qc.qualimap.main.NgsSmartMain.main(NgsSmartMain.java:103)
Tue Apr 28 08:56:52 EDT 2015 WARNING Cleanup output dir
Here is a sub-sample of the BAM file.
Thank you again
Java memory size is set to 1200M
Launching application...
QualiMap v.2.0
Built on 2014-08-28 17:03
Selected tool: bamqc
Available memory (Mb): 33
Max memory (Mb): 1118
Starting bam qc....
Loading sam header...
Loading locator...
Loading reference...
Number of windows: 400, effective number of windows: 304
Chunk of reads size: 1000
Number of threads: 12
Total processed windows:0
Number of reads: 1592618
Number of valid reads: 0
Number of duplicated reads: 0
Number of correct strand reads:0
Inside of regions...
Num mapped reads: 0
Num mapped first of pair: 0
Num mapped second of pair: 0
Num singletons: 0
Time taken to analyze reads: 5
Computing descriptors...
numberOfMappedBases: 0
referenceSize: 1215
numberOfSequencedBases: 0
numberOfAs: 0
Computing per chromosome statistics...
Failed to run bamqc
java.lang.IndexOutOfBoundsException: Index: 0, Size: 0
at java.util.ArrayList.rangeCheck(ArrayList.java:635)
at java.util.ArrayList.get(ArrayList.java:411)
at org.bioinfo.ngs.qc.qualimap.beans.BamStats.computeChromosomeStats(BamStats.java:1703)
at org.bioinfo.ngs.qc.qualimap.process.BamStatsAnalysis.run(BamStatsAnalysis.java:491)
at org.bioinfo.ngs.qc.qualimap.main.BamQcTool.execute(BamQcTool.java:197)
at org.bioinfo.ngs.qc.qualimap.main.NgsSmartTool.run(NgsSmartTool.java:187)
at org.bioinfo.ngs.qc.qualimap.main.NgsSmartMain.main(NgsSmartMain.java:103)
Tue Apr 28 08:56:52 EDT 2015 WARNING Cleanup output dir
Here is a sub-sample of the BAM file.
Thank you again
Konstantin Okonechnikov
Apr 30, 2015, 8:58:07 AM4/30/15
to qual...@googlegroups.com
Hi Antony,
thanks for sharing the data!
The problem was exactly because there were no mapped reads in the BAM file. Now the bug is fixed, additionally warning is reported if number of mapped reads equals to zero.
Here's the link to archive that includes the fix:
--
Konstantin
--
Antony Vincent
Apr 30, 2015, 2:35:11 PM4/30/15
to qual...@googlegroups.com
Thank you for the answer and the new build. Qualimap is really a nice tool.
Le lundi 27 avril 2015 14:04:40 UTC-4, Antony Vincent a écrit :
Reply all
Reply to author
Forward
0 new messages