Error when summarizing the mcc tree in TreeAnnotator v1.8.1
391 views
Skip to first unread message

Antonio
Dec 5, 2014, 4:01:08 PM12/5/14
to beast...@googlegroups.com
Dear all,
I'm working with a dataset of mitochondrial DNA from 308 different samples from a genera of mammals. I did a discrete phylogeographical analysis in BEAST 1.8.1 with chain lenght of MCMC of 200.000.000 generations, sampling every 5.000 steps.
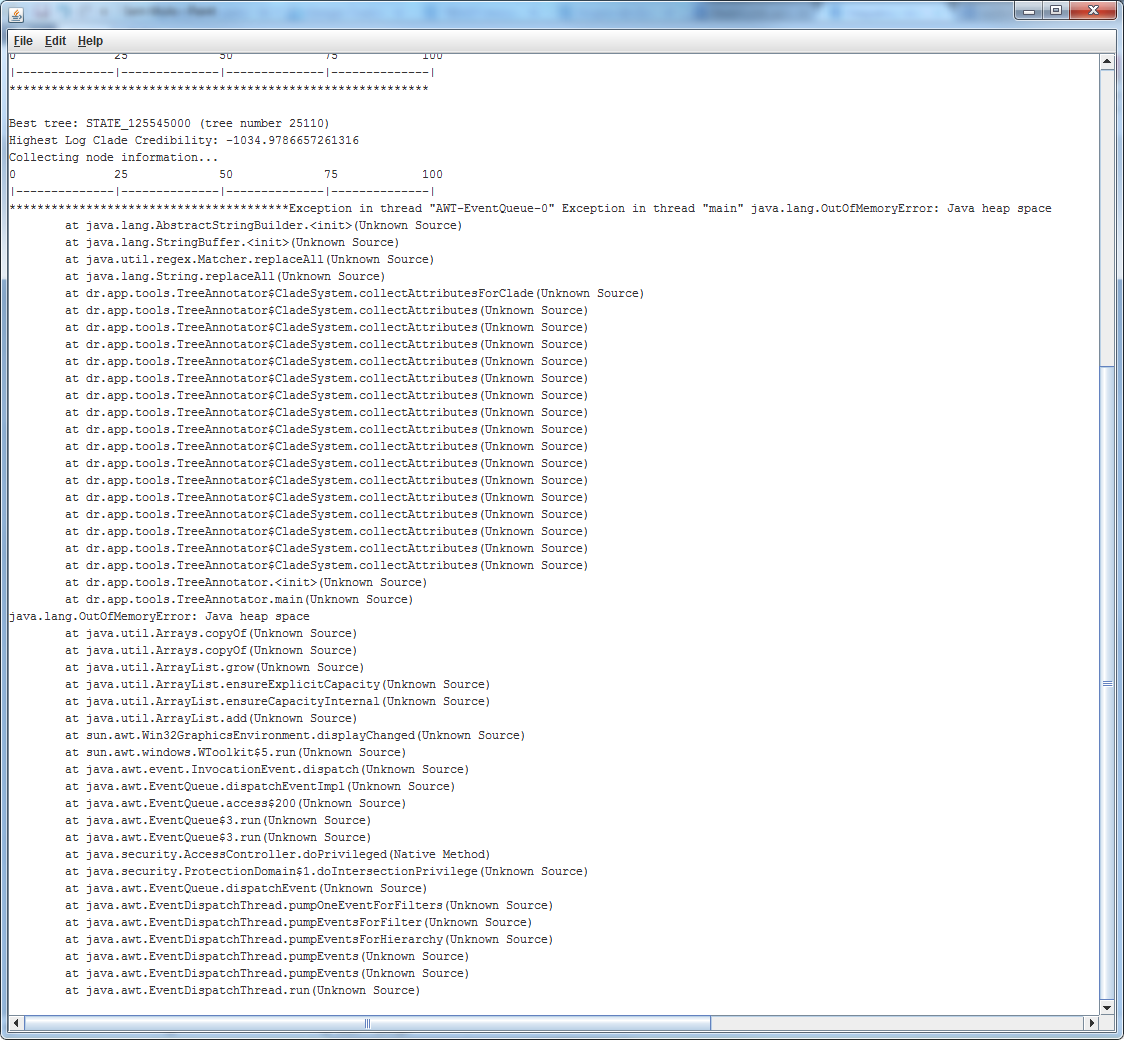
All my ESS values were very high and strong for all parameters. But when I tried to produce the MCC tree in TreeAnnotator software, I get an error like this (also see the figure Error1 attached):
Exception in thread "AWT-EventQueue-0" Exception in thread "main" java.lang.OutOfMemoryError: Java heap space
at java.lang.AbstractStringBuilder.<init>(Unknown Source)
at java.lang.StringBuffer.<init>(Unknown Source)
at java.util.regex.Matcher.replaceAll(Unknown Source)
at java.lang.String.replaceAll(Unknown Source)
at dr.app.tools.TreeAnnotator$CladeSystem.collectAttributesForClade(Unknown Source)
at dr.app.tools.TreeAnnotator$CladeSystem.collectAttributes(Unknown Source)
at dr.app.tools.TreeAnnotator$CladeSystem.collectAttributes(Unknown Source)
at dr.app.tools.TreeAnnotator$CladeSystem.collectAttributes(Unknown Source)
at dr.app.tools.TreeAnnotator$CladeSystem.collectAttributes(Unknown Source)
at dr.app.tools.TreeAnnotator$CladeSystem.collectAttributes(Unknown Source)
at dr.app.tools.TreeAnnotator$CladeSystem.collectAttributes(Unknown Source)
at dr.app.tools.TreeAnnotator$CladeSystem.collectAttributes(Unknown Source)
at dr.app.tools.TreeAnnotator$CladeSystem.collectAttributes(Unknown Source)
at dr.app.tools.TreeAnnotator$CladeSystem.collectAttributes(Unknown Source)
at dr.app.tools.TreeAnnotator$CladeSystem.collectAttributes(Unknown Source)
at dr.app.tools.TreeAnnotator$CladeSystem.collectAttributes(Unknown Source)
at dr.app.tools.TreeAnnotator$CladeSystem.collectAttributes(Unknown Source)
at dr.app.tools.TreeAnnotator$CladeSystem.collectAttributes(Unknown Source)
at dr.app.tools.TreeAnnotator$CladeSystem.collectAttributes(Unknown Source)
at dr.app.tools.TreeAnnotator$CladeSystem.collectAttributes(Unknown Source)
at dr.app.tools.TreeAnnotator$CladeSystem.collectAttributes(Unknown Source)
at dr.app.tools.TreeAnnotator.<init>(Unknown Source)
at dr.app.tools.TreeAnnotator.main(Unknown Source)
java.lang.OutOfMemoryError: Java heap space
at java.util.Arrays.copyOf(Unknown Source)
at java.util.Arrays.copyOf(Unknown Source)
at java.util.ArrayList.grow(Unknown Source)
at java.util.ArrayList.ensureExplicitCapacity(Unknown Source)
at java.util.ArrayList.ensureCapacityInternal(Unknown Source)
at java.util.ArrayList.add(Unknown Source)
at sun.awt.Win32GraphicsEnvironment.displayChanged(Unknown Source)
at sun.awt.windows.WToolkit$5.run(Unknown Source)
at java.awt.event.InvocationEvent.dispatch(Unknown Source)
at java.awt.EventQueue.dispatchEventImpl(Unknown Source)
at java.awt.EventQueue.access$200(Unknown Source)
at java.awt.EventQueue$3.run(Unknown Source)
at java.awt.EventQueue$3.run(Unknown Source)
at java.security.AccessController.doPrivileged(Native Method)
at java.security.ProtectionDomain$1.doIntersectionPrivilege(Unknown Source)
at java.awt.EventQueue.dispatchEvent(Unknown Source)
at java.awt.EventDispatchThread.pumpOneEventForFilters(Unknown Source)
at java.awt.EventDispatchThread.pumpEventsForFilter(Unknown Source)
at java.awt.EventDispatchThread.pumpEventsForHierarchy(Unknown Source)
at java.awt.EventDispatchThread.pumpEvents(Unknown Source)
at java.awt.EventDispatchThread.pumpEvents(Unknown Source)
at java.awt.EventDispatchThread.run(Unknown Source)
I did this different times and in other computers. I restarted my PC and tried again, but the same error was returned to me.
Curiously, everytime that I tried to sum the MCC tree, the error occurred in the same moment of the process: about 50+% of the process of collection information about the nodes.
Someone have an idea about this? Why is it occurs?
Someone have an idea about this? Why is it occurs?
I have another little doubts about the analysis that I'm doing.
In fact, I used an TN93+G site model with 4 gamma categories (JModeltest and Kakusan4 indicated it for me), and a asymmetric model with BSSVS. I have 14 different localities (states).
I used an uncorrelated lognormal relaxed molecular clock with ucld.mean with a gamma distribution with shape of 0.001 and scale of 1000 with initial value=10.
I used an uncorrelated lognormal relaxed molecular clock with ucld.mean with a gamma distribution with shape of 0.001 and scale of 1000 with initial value=10.
The ucld.stdev= default (exponential=0.333333). As suggested by Philippe, to estimate the overall state exchange rate, the prior location.clock.rate was set as
default (CTMC Rate Reference) since that I don't have an adequate prior information (Ferreira and Suchard, 2008).
I also estimated the root frequencies adding a frequencyModel in ancestralTreeLikelihood and sampling putting a deltaExchange operator:
<deltaExchange delta="0.01" weight="2">
<parameter idref="root.Freqs"/>
</deltaExchange>
My first question is about the estimation of divergence time. An work of 2012 estimated the age of the node that I'm interested now, by using some fossil calibrations in deep nodes.
I want to restrict the age of this node (tmrca prior) in 9.4 and 4.1, that were the HPD limit found by these authors. It's better to use a normal or uniform distribution? I know that using the normal
distribution I just need to put the mean (6.7, following the work of 2012) and choose a stdev that make the limits near to 9.4 and 4.1. But when I tried to test using a uniform distribution,
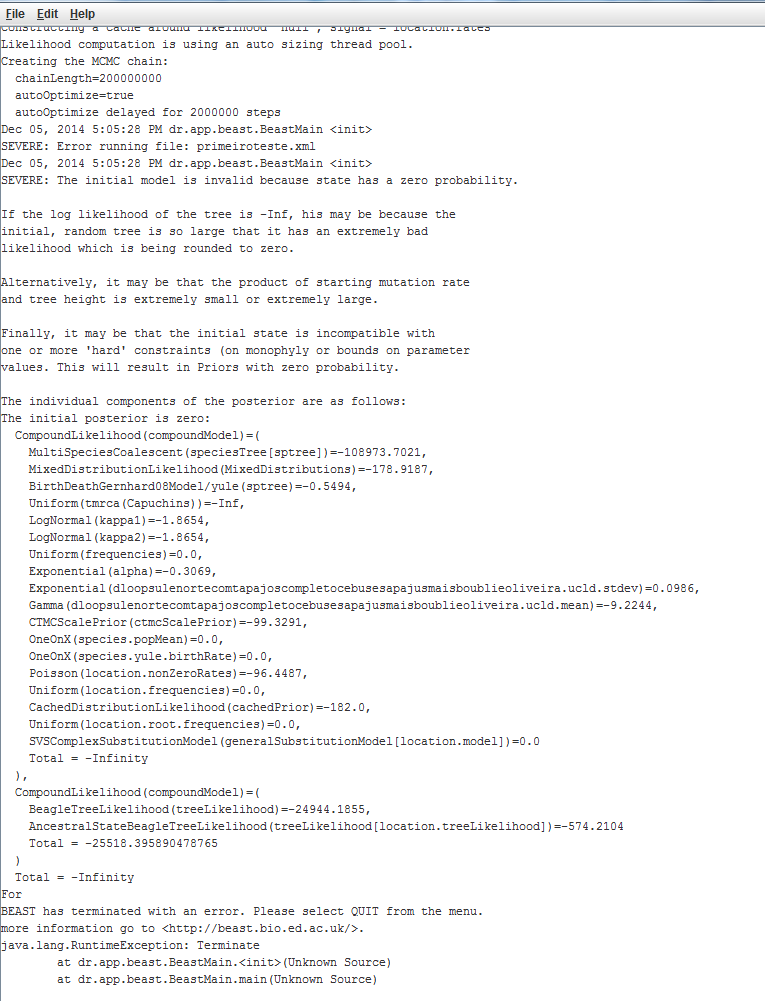
some error occurs. I put an initial value of 6.7 with an upper and lower values of 9.4 and 4.1, respectively. However, when I tried to run the BEAST v. 1.8.1 this error arises (figure Error 2):
Creating the MCMC chain:
chainLength=200000000
autoOptimize=true
autoOptimize delayed for 2000000 steps
Dec 05, 2014 5:05:28 PM dr.app.beast.BeastMain <init>
SEVERE: Error running file: primeiroteste.xml
Dec 05, 2014 5:05:28 PM dr.app.beast.BeastMain <init>
SEVERE: The initial model is invalid because state has a zero probability.
If the log likelihood of the tree is -Inf, his may be because the
initial, random tree is so large that it has an extremely bad
likelihood which is being rounded to zero.
Alternatively, it may be that the product of starting mutation rate
and tree height is extremely small or extremely large.
Finally, it may be that the initial state is incompatible with
one or more 'hard' constraints (on monophyly or bounds on parameter
values. This will result in Priors with zero probability.
The individual components of the posterior are as follows:
The initial posterior is zero:
CompoundLikelihood(compoundModel)=(
MultiSpeciesCoalescent(speciesTree[sptree])=-108973.7021,
MixedDistributionLikelihood(MixedDistributions)=-178.9187,
BirthDeathGernhard08Model/yule(sptree)=-0.5494,
Uniform(tmrca(Capuchins))=-Inf,
LogNormal(kappa1)=-1.8654,
LogNormal(kappa2)=-1.8654,
Uniform(frequencies)=0.0,
Exponential(alpha)=-0.3069,
Exponential(dloopsulenortecomtapajoscompletocebusesapajusmaisboublieoliveira.ucld.stdev)=0.0986,
Gamma(dloopsulenortecomtapajoscompletocebusesapajusmaisboublieoliveira.ucld.mean)=-9.2244,
CTMCScalePrior(ctmcScalePrior)=-99.3291,
OneOnX(species.popMean)=0.0,
OneOnX(species.yule.birthRate)=0.0,
Poisson(location.nonZeroRates)=-96.4487,
Uniform(location.frequencies)=0.0,
CachedDistributionLikelihood(cachedPrior)=-182.0,
Uniform(location.root.frequencies)=0.0,
SVSComplexSubstitutionModel(generalSubstitutionModel[location.model])=0.0
Total = -Infinity
),
CompoundLikelihood(compoundModel)=(
BeagleTreeLikelihood(treeLikelihood)=-24944.1855,
AncestralStateBeagleTreeLikelihood(treeLikelihood[location.treeLikelihood])=-574.2104
Total = -25518.395890478765
)
Total = -Infinity
For
BEAST has terminated with an error. Please select QUIT from the menu.
more information go to <http://beast.bio.ed.ac.uk/>.
java.lang.RuntimeException: Terminate
at dr.app.beast.BeastMain.<init>(Unknown Source)
at dr.app.beast.BeastMain.main(Unknown Source)
Why do this error occurs?
How can I put an uniforme distribution with the initial value of 6.7 and restricting the limit as 9.4 and 4.1 in this tmrca prior?
My second question is about the prior ucld.stdev. I set it as default (exponencial=0.333333), but is it the best to do? Is the uniform distribution better?
My last doubt is if I can do simultaneously the discrete phylogeographic and *BEAST analyzes estimating the divergence times at the same run in BEAST v. 1.8.1.
It will affect badly the run or not?
Thank you and Best Regards,
Antonio

{kind=link}
{kind=link}
mountainmanjared
Dec 6, 2014, 2:45:40 PM12/6/14
to beast...@googlegroups.com
Hi Antonio,
I'll just take a crack at your first issue. I would bet it's due to how many tree samples you have. 200,000,000/5,000 = 40,000 saved trees...which is a lot! There is a lot of information in each tree, too, given that you have 308 tips. I think you can take your .trees file into LogCombiner and thin the sampling rate by putting a value in the "Resample states at lower frequency" box. Your initial value was 5,000, so I'd put something on the order of 50,000 or more to cut it to 4,000 or fewer saved trees to bring into Tree Annotator.
Good luck!
Jared
I'll just take a crack at your first issue. I would bet it's due to how many tree samples you have. 200,000,000/5,000 = 40,000 saved trees...which is a lot! There is a lot of information in each tree, too, given that you have 308 tips. I think you can take your .trees file into LogCombiner and thin the sampling rate by putting a value in the "Resample states at lower frequency" box. Your initial value was 5,000, so I'd put something on the order of 50,000 or more to cut it to 4,000 or fewer saved trees to bring into Tree Annotator.
Good luck!
Jared
Antonio
Dec 6, 2014, 7:44:50 PM12/6/14
to beast...@googlegroups.com
Hi Jared,
Thank you for ur answer.
But I have some questions about your suggestion. I did a species tree analysis with this same chain length of MCMC (200.000.000) sampling every 5.000 steps.
But I have some questions about your suggestion. I did a species tree analysis with this same chain length of MCMC (200.000.000) sampling every 5.000 steps.
And, in this case, I have produced a summarized tree. It because the amount of information in the case of the species tree analysis is lower?
If I do what you are suggesting me, I will not lose too much information? I know that if the sampling of parameters in log files is not too much frequent, the information that we get by the run of MCMC may be not so good. Do you think that only 4.000 trees (or samples of parameters) is good to get good estimatives?
There are other way to solve this problem? If I use a more strong computer can I do the summarization of 36.000 trees?
If I do what you are suggesting me, I will not lose too much information? I know that if the sampling of parameters in log files is not too much frequent, the information that we get by the run of MCMC may be not so good. Do you think that only 4.000 trees (or samples of parameters) is good to get good estimatives?
There are other way to solve this problem? If I use a more strong computer can I do the summarization of 36.000 trees?
Thank you again,
Antonio
mountainmanjared
Dec 6, 2014, 8:45:37 PM12/6/14
to beast...@googlegroups.com
Hi Antonio,
I would suspect that Tree Annotator was able to summarize your species tree posterior because it had fewer tips than 308? And yes, the phylogeographical analysis has a lot more information contained in the trees (diffusion rate, location, etc.) than even a normal gene tree analysis with the same number of tips. But I can only guess. Similarly, I don't know if a more "powerful" computer would be able to solve your problem. I have not tried that, I've just decreased the sampling rate.
I don't know the literature, but I think 4,000 trees should be more than ample to give a good summary of your posterior. I know that 5000 works for me, even 1,000 often times. My suggestion would be to try what I suggested before, and see how your ESS values look with 4,000 samples.
Good luck,
Jared
I would suspect that Tree Annotator was able to summarize your species tree posterior because it had fewer tips than 308? And yes, the phylogeographical analysis has a lot more information contained in the trees (diffusion rate, location, etc.) than even a normal gene tree analysis with the same number of tips. But I can only guess. Similarly, I don't know if a more "powerful" computer would be able to solve your problem. I have not tried that, I've just decreased the sampling rate.
I don't know the literature, but I think 4,000 trees should be more than ample to give a good summary of your posterior. I know that 5000 works for me, even 1,000 often times. My suggestion would be to try what I suggested before, and see how your ESS values look with 4,000 samples.
Good luck,
Jared
Antonio Marcio Gomes Martins Junior
Dec 10, 2014, 10:07:35 AM12/10/14
to beast...@googlegroups.com
Thank you Jared.
I did it and the ESS values and the values of parameters didn't change (or statistically speaking).
Now I could sum the .trees file.
I did it and the ESS values and the values of parameters didn't change (or statistically speaking).
Now I could sum the .trees file.
Best,
Antonio
--
You received this message because you are subscribed to the Google Groups "beast-users" group.
To unsubscribe from this group and stop receiving emails from it, send an email to beast-users...@googlegroups.com.
To post to this group, send email to beast...@googlegroups.com.
Visit this group at http://groups.google.com/group/beast-users.
For more options, visit https://groups.google.com/d/optout.
Reply all
Reply to author
Forward
0 new messages