LSV detected but STAR see no reads mapped to the corresponding gene

Elsa Claude
Elsa Claude

Jordi Vaquero
Hello Elsa,
The question falls in “how do you find which read belongs to each gene”. Majiq uses a set of rules, like if the junctions is anotated or not anotated but connects known splicesites, or is the only gene that may have that region, and so on.
If you check genome browser for that gene there are several genes in the region. It might happen that some reads describe de novo junctions in the gene or in another overlaping gene, majiq uses the set of euristics to find the most probable one. Other tools migh discard those reads or map them in a diferent gene.
I am not sure who the gene count from star Works in that sense.
I hope this answers your question
Jordi Vaquero
De: Elsa Claude
Enviat: Thursday, April 22, 2021 1:28 PM
Per a: majiq_voila
Tema: [Majiq] Re: LSV detected but STAR see no reads mapped to the corresponding gene
Hi,
Does anyone have a possible explanation? ;)
Cheers,
Elsa
Le mardi 9 mars 2021 à 12:17:50 UTC+1, Elsa Claude a écrit :
Hello,
I think the answer may be pretty simple and I must be missing an important detail but... I don't understand how is that possible that MAJIQ can find an LSV when STAR (which generated the bam files I'm using for the MAJIQ analysis) -quantmode Genecounts paramater gives an output where it seems that no 'uniquely' read is mapped to the gene.
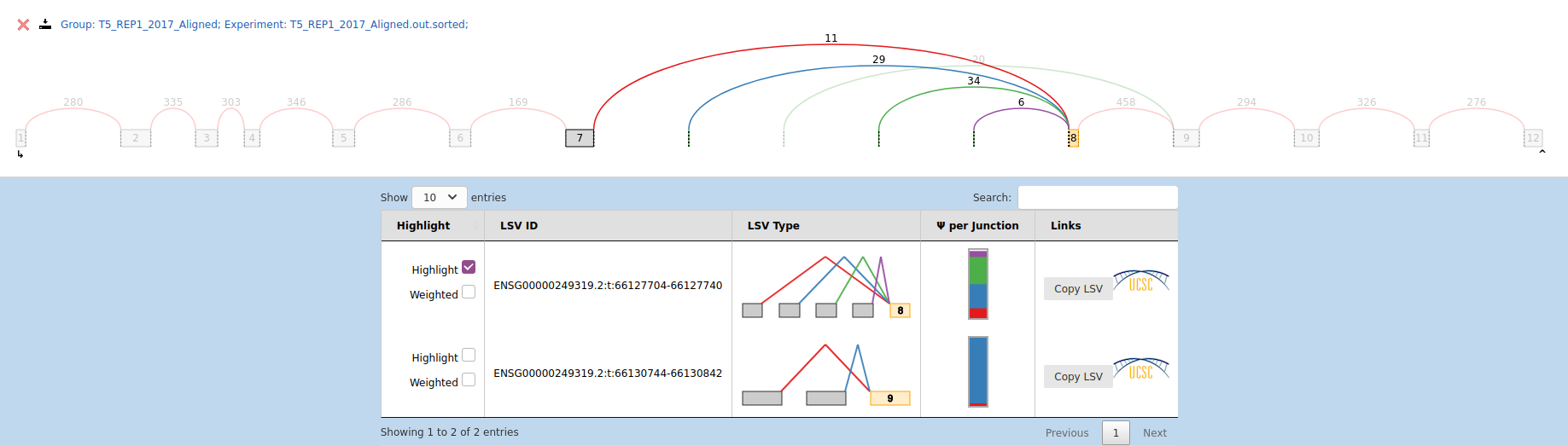
Here is an example of an LSV in this situation :
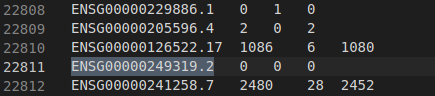
And here is the count of reads for the gene involved, and output by STAR using the GeneCount parameter :
Among the 600 LSVs (around) I am interested in, only 9 genes involved have this kind of issues. It's a minority but I'm still interested in knowing what is the explanation.
Thanks for your time,
Have a nice day,
Elsa
--
You received this message because you are subscribed to the Google Groups "majiq_voila" group.
To unsubscribe from this group and stop receiving emails from it, send an email to majiq_voila...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/majiq_voila/fb31f206-62c5-4b78-ad69-4610faf31a19n%40googlegroups.com.
Joseph Aicher
- STAR uses htseq-count defaults. This only counts reads that are:
- uniquely aligned reads,
- to genomic regions that belong unambiguously to exactly one gene
- MAJIQ counts uniquely aligned reads
- but if a junction is shared by multiple genes' transcripts, we can't differentiate which gene it belongs to
- we have the choice of (a) pretending that the junction doesn't exist or (b) considering it as part of multiple genes
- MAJIQ chooses (b), which we think is more appropriate for splicing quantification
- If we look at the Ensembl genome browser at the gene you've linked (https://useast.ensembl.org/Homo_sapiens/Gene/Summary?db=core;g=ENSG00000249319;r=7:66087761-66152277;t=ENST00000450043), we can see that the exons fully overlap other genes
- so STAR/htseq-count will not count any reads that map to the gene because they share the same genomic location with other genes. The fact that your data are stranded doesn't even help because the genes are oriented in the same direction
To view this discussion on the web visit https://groups.google.com/d/msgid/majiq_voila/C9693115-7C60-42BE-A9A0-30A15FA62CB6%40hxcore.ol.