GeneRax SpeciesEvents Counts
139 views
Skip to first unread message

Clifton Lewis
Jun 23, 2021, 2:22:36 PM6/23/21
to GeneRax
Hi Benoit,
I was just wondering what the difference in the per_species_event_counts.txt in the main output folder in comparison to the one in the reconciliation folder is. Does the S= speciation; SL= Speciation loss; D=duplication; T= Transfers; TL=Transfer losses.? Could you possibly just explain the calculation logic used for this and how this is different to the eventscounts output in the reconciliation folder? I may have missed some straightforward thing on the github page, if so apologies. Appreciate your help in this.
Thanks,
Clifton

Benoit Morel
Jun 24, 2021, 3:39:25 AM6/24/21
to GeneRax
Hi Clifton,
Thank you for the suggestion, this is indeed missing in the wiki. I just added a section here: https://github.com/BenoitMorel/GeneRax/wiki/GeneRax#reconciliation-events
Please let me know if this answers your questions.
Also, I plan to reorganize the wiki and to add figures, but I'll need more time for that :-)
Best,
Benoit
Clifton Lewis
Jun 24, 2021, 6:04:37 AM6/24/21
to Benoit Morel, GeneRax
Hi Benoit,
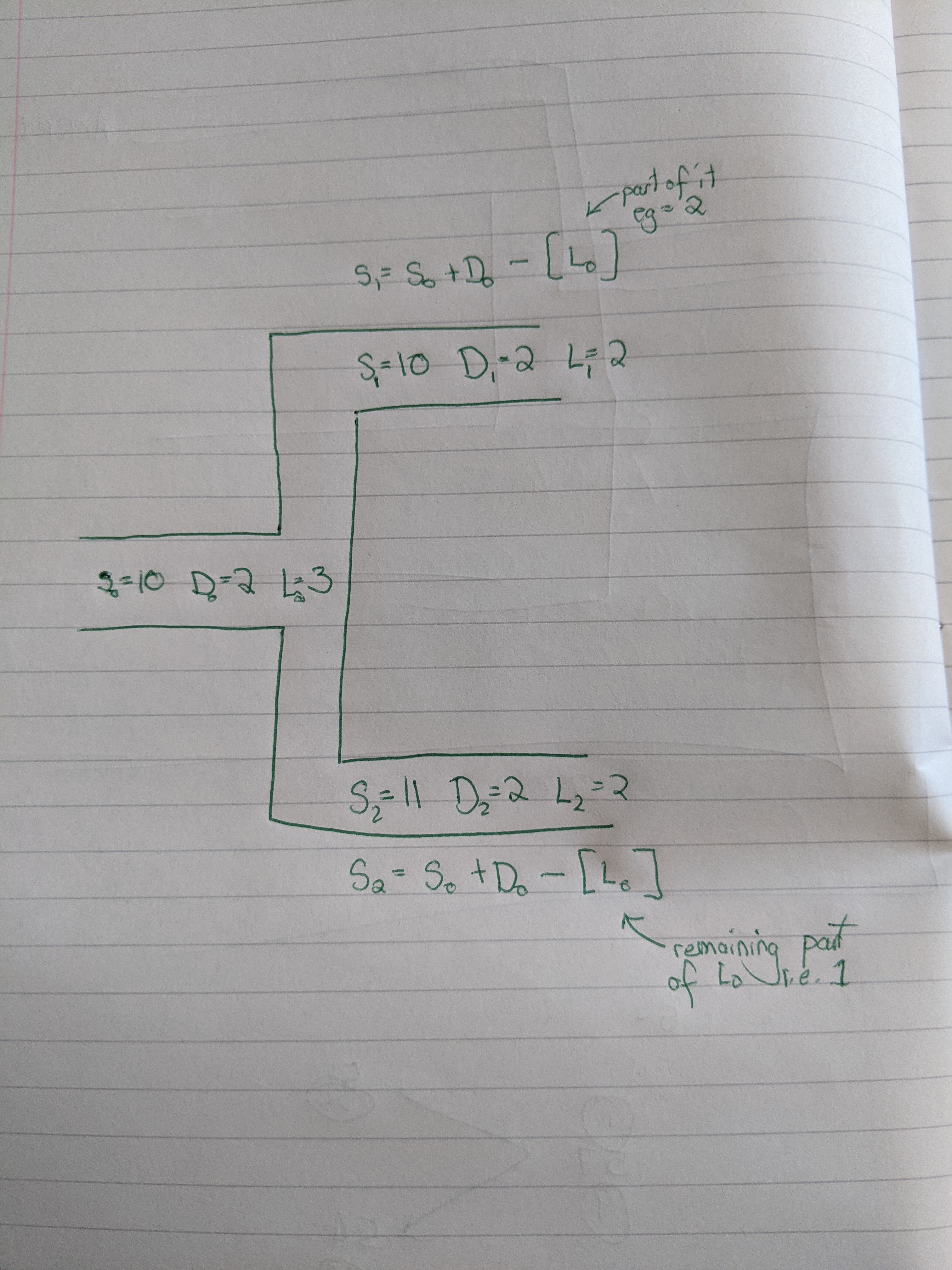
Thank you for the explanation, it really helps but just so I have it understood correctly the speciation count is based on the number of speciation at the previous node plus the number of duplications and some of the speciation loss while the other split from the previous node accounts for the remaining losses? I have attached a simple sketch of what I think the calculation logic is, I was wondering if you could tell if I have assumed something incorrectly. Appreciate your help again.
Thanks,
Clifton
--
You received this message because you are subscribed to a topic in the Google Groups "GeneRax" group.
To unsubscribe from this topic, visit https://groups.google.com/d/topic/generaxusers/yC_QQS9fFDY/unsubscribe.
To unsubscribe from this group and all its topics, send an email to generaxusers...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/generaxusers/350ea689-ff58-4214-b2f1-1bb0769f4b43n%40googlegroups.com.
For more options, visit https://groups.google.com/d/optout.
Benoit Morel
Jun 24, 2021, 6:25:34 AM6/24/21
to Clifton Lewis, GeneRax
Your reasoning looks correct to me. However, gene trees do not have to originate from the species root node. As a consequence, you might have more genes than expected when you go doing the species tree.
Benoit Morel
Jun 24, 2021, 6:29:20 AM6/24/21
to Clifton Lewis, GeneRax
I meant "when you go down" :-)
Clifton Lewis
Jun 24, 2021, 8:05:25 AM6/24/21
to Benoit Morel, GeneRax
Hi Benoit,
That's interesting because I just ran some analysis on my own data with each family run as it's own reconciliation run. I then totaled up the speciation duplication and losses and the numbers dont seem to add up based on the prior calculation logic as for example for one of the nodes I have S=23; D=6; L=6 and the daughter nodes are S=24; D=5; L=6 and S=23; D=2; L=3 respectively. I can't seem to figure out why the numbers dont add up? Am I missing something really basic here? Should I have run it all in a single generax run. i.e. I can't compare numbers across generax runs?
Thanks,
Clifton
Benoit Morel
Jun 25, 2021, 4:38:51 AM6/25/21
to Clifton Lewis, GeneRax
Hi Clifton,
Can you just tell me what is L in your previous text? Is it the number of SL events attached to this species? Does S represent the number of S+SL or just S without SL?
Here is also something that was maybe unclear: for a given (potentially ancestral) species, the duplications happen along the branch BEFORE the speciation events. Let's take an example without any loss event to make it simpler. Let us assume that in the parent node P, you have D=6 and S=10. Then after the speciation, there will be 10 genes in each new child lineage C1 and C2. Then if C1 has 4 duplications (D=4), it should end up with S=14.
In addition, some gene trees could be rooted at C1, and thus not appear in P.
I'll look again at your example once I understand what L and S exactly represent :-)
Benoit
Benoit Morel
Jun 28, 2021, 3:58:25 AM6/28/21
to GeneRax
Hi Clifton,
Back to your example, with P being the parent species and C1 and C2 its children.
P: S=23; D=6; L=6
C1: S=24; D=5; L=6
C2: S=23; D=2; L=3
When P speciates, there are 23 gene copies in its genome (S=23). D=6 is irrelevant for the children, because the duplications happened before the speciation (the number S=23 is a consequence of the 6 speciations). L=6 means that there are 6 losses to distribute between C1 and C2 (which is counter-intuitive and that I maybe want to change in a next release). In this example, 4 out of the 6 losses will happen along C1, and 2 along C2.
Right after P speciates, C1 has 23 gene copies. Then
there are 5 duplications (D=5 for C1) and 4 losses (4 out of the 6
that are indicated in P). This results in 23+5-4=24 gene copies when C1
speciates (S=24 for C1 ok!)
Right after P speciates, C2 has 23 gene copies. Then
there are 2 duplications (D=2 for C2) and 2 losses (2 out of the 6
that are indicated in P). This results in 23+2-2=23 gene copies when C1
speciates (S=23 for C2 ok!)
This indeed deserves some explanation in the wiki... But as I said, I'll need more time, because I think this will be better with figures.
I hope this answer your questions
Best,
Benoit
Benoit Morel
Jun 28, 2021, 4:01:39 AM6/28/21
to GeneRax
Also, regarding your screenshot:
S1= S0 + D0 - [L0]
should then be
S1= S0 + D1 - [L0]
Sorry that I haven't spotted that earlier.
Clifton Lewis
Jun 30, 2021, 4:55:21 AM6/30/21
to Benoit Morel, GeneRax
Hi Benoit,
Thank you for this. I went back over my results and the numbers make sense now. It is a bit less intuitive better I get the logic now that I have your great explanation . I think it would actually be lovely to have figures in the wiki to help explain the logic. Thank you for all your help in explaining this to me and your patience.
Thanks,
Clifton
To view this discussion on the web visit https://groups.google.com/d/msgid/generaxusers/129e92a2-04fd-4829-9235-bfaba5bd92b7n%40googlegroups.com.

{kind=link}
Bin He
May 18, 2022, 9:11:58 AM5/18/22
to GeneRax
Hi Benoit,
I want to follow up on Clifton's question as I, too, struggled to understand how speciation-loss is calculated. Your example above now made it clear, but am I correct in saying that how the losses in P are distributed in C1 and C2 are not explicitly stated (4 and 2 are given by you in the example). In that case, we will need to work out the numbers by following the calculations below?
In practice, I would like to show the number of inferred duplications and the number of extinction events along each branch. So in the above example, I feel the intuitive way to show the results would be to represent 4 and 2 as the number of extinction events along the C1 and C2 branches. This also seems to be how ThirdKind and redPhyloVisu represent them (with "x"s along the child branches).
What's your thought on this?
Thanks
Bin
Benoit Morel
May 19, 2022, 3:59:59 AM5/19/22
to GeneRax
Hi Bin,
Yes you are right about everything. I just opened an issue in our github (https://github.com/BenoitMorel/GeneRax/issues/59) regarding this issue of not having the extinctions explicitly counted in the species in which they happen. I will try to fix this asap or to provide a bypass to get the extinction counts without having to do convoluted operations. As you said, what we should have is the losses as represented in ThirdKind.
Best,
Benoit
Bin He
May 22, 2022, 4:53:21 PM5/22/22
to GeneRax
Thanks Benoit for the quick response! Really appreciate it! -- Bin
Reply all
Reply to author
Forward
0 new messages