Calculate Ar-MoS2 potential energy
58 views
Skip to first unread message

toto Qian
Jan 2, 2020, 7:56:48 AM1/2/20
to cp2k
Dear cp2k developers and users,
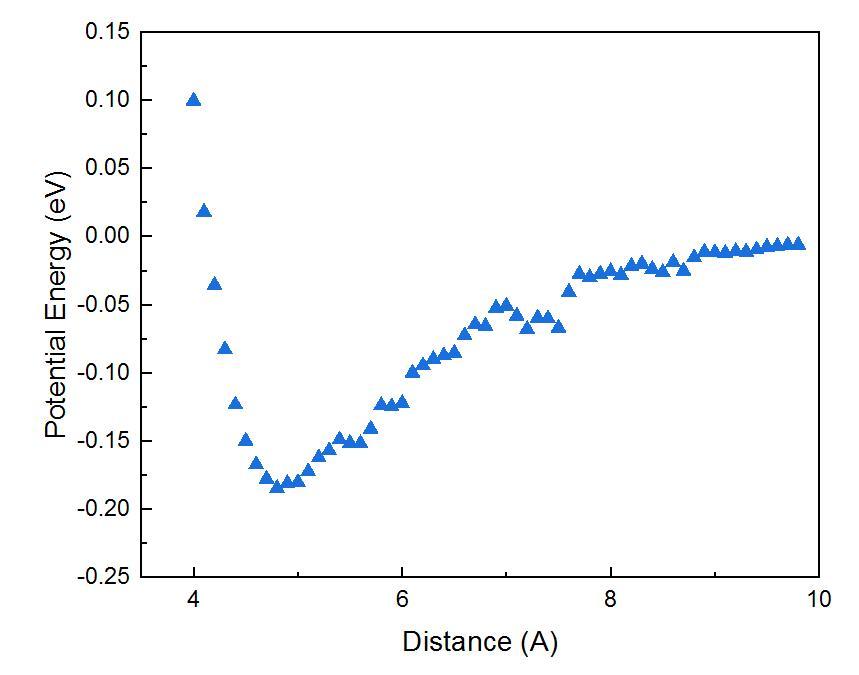
I am trying to calculate Ar-MoS2(2D) potential energy (= Energy_Ar&MoS2 - Energy_Ar - Energy_MoS2), while the potential energy trend is not smooth, Fig. How can I solve it? Or is it just normal?
I used def2-TZVP, DFTD3, and pseudopotential for Mo atom. The SCF convergence set is: EPS_SCF 1.0E-7 and OUTER_SCF EPS_SCF 2.0E-7
Happy new Decade.
Message has been deleted

Thomas Kühne
Jan 4, 2020, 11:41:02 AM1/4/20
to cp...@googlegroups.com
Dear Toto Qian,
you may try tightening EPS_DEFAULT. Also please make sure
that your SCF has always properly converged MAX_SCF of the
innrer loop appears a little bit low to me in particular if you are
using different EPS_SCF values (there is not much reason to
do so anyhow).
Cheers,
Thomas
Am 02.01.2020 um 13:56 schrieb toto Qian <qjh103...@gmail.com>:
Dear cp2k developers and users,I am trying to calculate Ar-MoS2(2D) potential energy (= Energy_Ar&MoS2 - Energy_Ar - Energy_MoS2), while the potential energy trend is not smooth, Fig. How can I solve it? Or is it just normal?I used def2-TZVP, DFTD3, and pseudopotential for Mo atom. The SCF convergence set is: EPS_SCF 1.0E-7 and OUTER_SCF EPS_SCF 2.0E-7
<QQ截图20200102204426.jpg>
Happy new Decade.--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/e6591a41-8d78-4602-a965-fea4112abf54%40googlegroups.com.
<QQ截图20200102204426.jpg>
==============================
Thomas D. Kühne
Dynamics of Condensed Matter
Chair of Theoretical Chemistry
University of Paderborn
Warburger Str. 100
D-33098 Paderborn
Germany
toto Qian
Jan 5, 2020, 8:18:29 PM1/5/20
to cp2k
Dear Thomas,
Thank you for your suggestion. I tightened EPS_DEFAULT from 1E-10 to 1E-14 and the distance-potential trend is smooth now! The MAX_SCF is enough for my calculations. So I did not modify it.
Best Regards,
Toto Qian
在 2020年1月5日星期日 UTC+8上午12:41:02,tkuehne写道:
在 2020年1月5日星期日 UTC+8上午12:41:02,tkuehne写道:
Dear Toto Qian,you may try tightening EPS_DEFAULT. Also please make surethat your SCF has always properly converged MAX_SCF of theinnrer loop appears a little bit low to me in particular if you areusing different EPS_SCF values (there is not much reason todo so anyhow).Cheers,Thomas
Am 02.01.2020 um 13:56 schrieb toto Qian <qjh103...@gmail.com>:
Dear cp2k developers and users,I am trying to calculate Ar-MoS2(2D) potential energy (= Energy_Ar&MoS2 - Energy_Ar - Energy_MoS2), while the potential energy trend is not smooth, Fig. How can I solve it? Or is it just normal?I used def2-TZVP, DFTD3, and pseudopotential for Mo atom. The SCF convergence set is: EPS_SCF 1.0E-7 and OUTER_SCF EPS_SCF 2.0E-7<QQ截图20200102204426.jpg>
Happy new Decade.--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/e6591a41-8d78-4602-a965-fea4112abf54%40googlegroups.com.
<QQ截图20200102204426.jpg>
Reply all
Reply to author
Forward
0 new messages