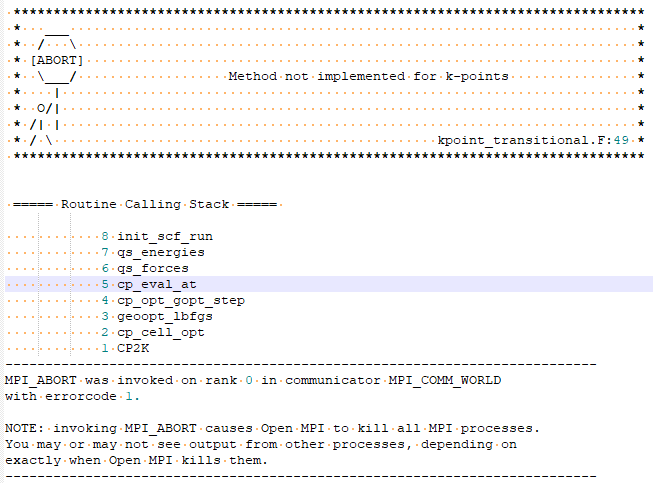
I would like to perform a DFT+U calculation with LaFeO3 perovskite. However, I have met the following error for KPOINTS setting and I am not sure where is the problem from. Could you kindly help me with this? It would be significantly appreciated for your help.
Another question is that is it ok to set two BASIS_SET_FILE_NAME within one input file, as I checked that there is no basis set for La in the BASIS_MOLOPT file.
For your reference, I run this calculation on my laptop with cp2k.popt version 5.1. I also attach the input file here for your information. Thanks.
!*************************GLOBAL SETTINGS**********************************
&GLOBAL
PRINT_LEVEL LOW
PROJECT_NAME lfo_rlx
RUN_TYPE CELL_OPT
SAVE_MEM T
&END GLOBAL
!*************************MOTION SETTINGS**********************************
&MOTION
&CELL_OPT
OPTIMIZER LBFGS
TYPE DIRECT_CELL_OPT
&END CELL_OPT
&END MOTION
!************************FORCE_EVAL SETTINGS********************************
&FORCE_EVAL
STRESS_TENSOR ANALYTICAL
METHOD QS
!********************FORCE_EVAL -> DFT SETTINGS*****************************
&DFT
BASIS_SET_FILE_NAME BASIS_MOLOPT_UCL
BASIS_SET_FILE_NAME BASIS_MOLOPT
POTENTIAL_FILE_NAME GTH_POTENTIALS
UKS T
CHARGE 0
PLUS_U_METHOD MULLIKEN
!****************FORCE_EVAL -> DFT -> SCF SETTINGS**************************
&SCF
ADDED_MOS 100
MAX_SCF 200
EPS_SCF 1.0E-6
SCF_GUESS RESTART
&DIAGONALIZATION T
ALGORITHM STANDARD
&END DIAGONALIZATION
&OUTER_SCF
EPS_SCF 1.0E-5
MAX_SCF 5
&END OUTER_SCF
&SMEAR T
METHOD FERMI_DIRAC
ELECTRONIC_TEMPERATURE [K] 300
&END SMEAR
&MIXING T
METHOD PULAY_MIXING
ALPHA 0.1
BETA [bohr^-1] 0.001
NMIXING 5
NBUFFER 8
&END MIXING
&END SCF
!****************FORCE_EVAL -> DFT -> QS SETTINGS***************************
&QS
EPS_DEFAULT 1.0E-12
EXTRAPOLATION USE_GUESS
METHOD GAPW
&END QS
!***************FORCE_EVAL -> DFT -> GRID SETTINGS**************************
&MGRID
NGRIDS 5
CUTOFF [Ry] 600
REL_CUTOFF [Ry] 60
&END MGRID
!*****************FORCE_EVAL -> DFT -> XC SETTINGS**************************
&XC
FUNCTIONAL_ROUTINE NEW
&XC_FUNCTIONAL PBE
&END XC_FUNCTIONAL
&END XC
!***************FORCE_EVAL -> DFT -> POISSON SETTINGS************************
&POISSON
POISSON_SOLVER PERIODIC
PERIODIC XYZ
&END POISSON
!***************FORCE_EVAL -> DFT -> KPOINTS SETTINGS************************
&KPOINTS
SCHEME MONKHORST-PACK 2 2 2
FULL_GRID .TRUE.
&END KPOINTS
&END DFT
!********************FORCE_EVAL -> SUBSYS SETTINGS***************************
&SUBSYS
&CELL
ABC [angstrom] 7.91435 7.91435 7.91435
ALPHA_BETA_GAMMA [deg] 90 90 90
PERIODIC XYZ
&END CELL
&KIND La
ELEMENT La
BASIS_SET DZVP-MOLOPT-SR-GTH
POTENTIAL GTH-PBE-q11
&END KIND
&KIND Fe
ELEMENT Fe
BASIS_SET DZVP-MOLOPT-SR-GTH
POTENTIAL GTH-PBE-q16
MAGNETIZATION 2.0
&DFT_PLUS_U T
L 2
U_MINUS_J [eV] 5.3
&END DFT_PLUS_U
&END KIND
&KIND O
ELEMENT O
BASIS_SET DZVP-MOLOPT-SR-GTH
POTENTIAL GTH-PBE-q6
&END KIND
&TOPOLOGY
COORD_FILE_FORMAT XYZ
&END TOPOLOGY
&END SUBSYS
!********************FORCE_EVAL -> PRINT SETTINGS****************************
&PRINT
&FORCES ON
&END FORCES
&END PRINT
&END FORCE_EVAL


{kind=link}