Convergence failure and incorrect solvation energy with SCCS in cp2k-2022.1

Wiko Ann

Krack Matthias (PSI)
Hi Wiko
- Slab: SCCS in CP2K is not implemented for k points and not yet tested with smearing. I guess that you are using one of these features for the Ag(111) slab which might cause problems.
- H3O+: The results are sensible to the choice of the parameters RHO_MIN and RHO_MAX. Their values differ especially for neutral and ionic systems. What do you get for the electrostatic part of the solvation energy?
Just as minor improvements (do not help for the issue above), I suggest to use for H3O+ the default diagonalization instead of OT (i.e. &OT off) and a smaller EPS_DEFAULT value (at least 1.0E-12 instead of 1.0E-10).
Best
Matthias
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to
cp2k+uns...@googlegroups.com.
To view this discussion on the web visit
https://groups.google.com/d/msgid/cp2k/0420c53c-f0bb-49dc-85fa-2124328053d8n%40googlegroups.com.
Krack Matthias (PSI)
PS: Do not use with SCCS “PSOLVER MT” and “PERIODIC NONE” but the defaults.
M.
From: cp...@googlegroups.com <cp...@googlegroups.com> on behalf of Krack Matthias (PSI) <matthia...@psi.ch>
Date: Monday, 7 November 2022 at 16:53
To: cp...@googlegroups.com <cp...@googlegroups.com>
Subject: Re: [CP2K:17989] Re: Convergence failure and incorrect solvation energy with SCCS in cp2k-2022.1
Hi Wiko
1. Slab: SCCS in CP2K is not implemented for k points and not yet tested with smearing. I guess that you are using one of these features for the Ag(111) slab which might cause problems.
2. H3O+: The results are sensible to the choice of the parameters RHO_MIN and RHO_MAX. Their values differ especially for neutral and ionic systems. What do you get for the electrostatic part of the solvation energy?
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZRAP278MB08274F9BF3607A244882B3C3F43C9%40ZRAP278MB0827.CHEP278.PROD.OUTLOOK.COM.
Wiko Ann
Wiko Ann
Krack Matthias (PSI)
Hi Wiko
- That’s indeed a problem.
- That’s correct, SCCS is meant to work with POISSON_SOLVER periodic and PERIODIC xyz.
Best
Matthias
From: cp...@googlegroups.com <cp...@googlegroups.com> on behalf of Wiko Ann <toda...@gmail.com>
Date: Tuesday, 8 November 2022 at 03:44
To: cp2k <cp...@googlegroups.com>
Subject: Re: [CP2K:17992] Re: Convergence failure and incorrect solvation energy with SCCS in cp2k-2022.1
Dear Matthias:
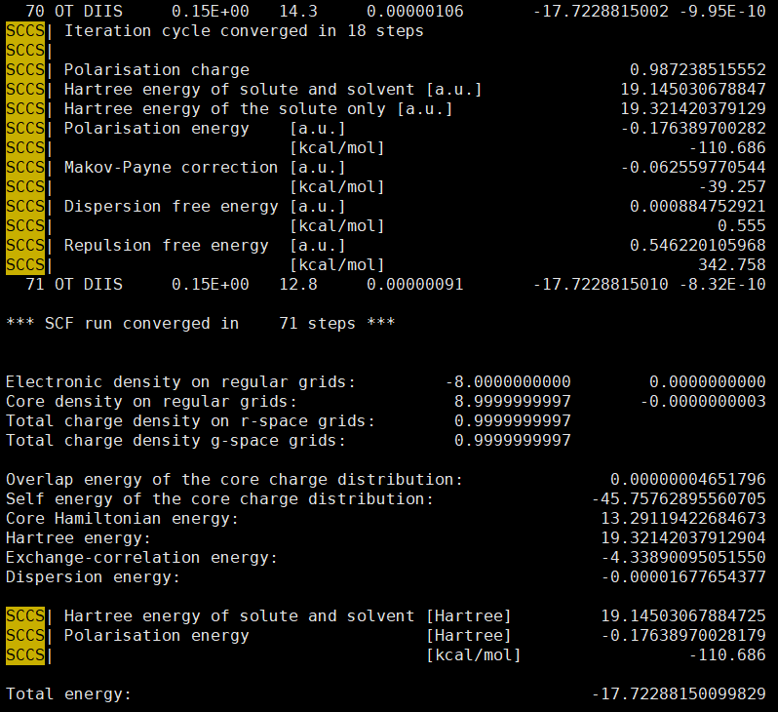
Thanks a lot for your reply. The electrostatic energy is posted below.
1. I did use k points and smearing in the calculation of slab. It should be the problem.
2. Did you mean if I want to obtain the solvation energy of H3O+, I can use “PSOLVER MT” and “PERIODIC NONE” to calculate cluster energy in gas phase, and then use the default setting when applying SCCS?
Regards
Wiko

To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ea085a09-02d3-425f-ac2c-cfcf2f1564e6n%40googlegroups.com.

Anton Lytvynenko
Dear Krack, dear all,
I'd like to ask another related question -- if SCCS is capable to model water without addition of some explicit water molecules in such situation?
Yours,
Anton
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZRAP278MB0827413D164197DF78B9DC76F43F9%40ZRAP278MB0827.CHEP278.PROD.OUTLOOK.COM.
Krack Matthias (PSI)
The box is not charged with the defaults “POISSON_SOLVER periodic” and “PERIODIC xyz” due to the automatically added neutralizing background charge. The periodic box cannot be charged in that case as this would add an infinite interaction energy term. In any case, the box should be chosen large enough like for gas phase calculations without periodicity.
Charged systems like H3O+ need special care and additional correction terms are required. Currently, CP2K provides only an approximative correction (Makov and Payne) for such systems.
Matthias
From: cp...@googlegroups.com <cp...@googlegroups.com> on behalf of Wiko Ann <toda...@gmail.com>
Date: Tuesday, 8 November 2022 at 04:13
To: cp2k <cp...@googlegroups.com>
Subject: Re: [CP2K:17993] Re: Convergence failure and incorrect solvation energy with SCCS in cp2k-2022.1
And if I use the default setting in gas phase, Will the additional interaction between charged periodic boxes be included in the total energy?
在2022年11月8日星期二 UTC+8 10:44:23<Wiko Ann> 写道:
Dear Matthias:
Thanks a lot for your reply. The electrostatic energy is posted below.
1. I did use k points and smearing in the calculation of slab. It should be the problem.
2. Did you mean if I want to obtain the solvation energy of H3O+, I can use “PSOLVER MT” and “PERIODIC NONE” to calculate cluster energy in gas phase, and then use the default setting when applying SCCS?
Regards
Wiko
.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/d4cca932-39ee-47ac-8a78-3dbf619d0588n%40googlegroups.com.
Wiko Ann
Krack Matthias (PSI)
Hi Anton
You are free to add explicit water molecules when using SCCS, but that’s not required, though it might be better to have some explicit water.
Best
Matthias
From: cp...@googlegroups.com <cp...@googlegroups.com> on behalf of Anton Lytvynenko <anton.s.l...@gmail.com>
Date: Tuesday, 8 November 2022 at 10:28
To: cp...@googlegroups.com <cp...@googlegroups.com>
Subject: Re: [CP2K:17995] Re: Convergence failure and incorrect solvation energy with SCCS in cp2k-2022.1
Dear Krack, dear all,
I'd like to ask another related question -- if SCCS is capable to model water without addition of some explicit water molecules in such situation?
Yours,
Anton
08.11.2022 10:17, Krack Matthias (PSI) пише:
Hi Wiko
1. That’s indeed a problem.
2. That’s correct, SCCS is meant to work with POISSON_SOLVER periodic and PERIODIC xyz.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/37c2571e-99f6-0535-d8e7-e8d3813118c8%40gmail.com.
Anton Lytvynenko
Dear Matthias,
thank your very much for the explanation.
Yours,
Anton
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZRAP278MB082703DA579071D939E44035F43E9%40ZRAP278MB0827.CHEP278.PROD.OUTLOOK.COM.
Krack Matthias (PSI)
Hi Wiko
After running checks using SCCS with POISSON_SOLVER MT and “PERIODIC none” for charged and uncharged molecules (solutes), I have correct my previous comment. CP2K/SCCS with MT seems to work fine which makes it the suggested setup especially for charged systems with SCCS. Note, that you need relatively high cutoff values (>800 Ry) and large cells for numerically accurate forces.
Sorry for confusing
Matthias
From: cp...@googlegroups.com <cp...@googlegroups.com> on behalf of Krack Matthias (PSI) <matthia...@psi.ch>
Date: Tuesday, 8 November 2022 at 10:17
To: cp...@googlegroups.com <cp...@googlegroups.com>
Subject: Re: [CP2K:17994] Re: Convergence failure and incorrect solvation energy with SCCS in cp2k-2022.1
Hi Wiko
1. That’s indeed a problem.
2. That’s correct, SCCS is meant to work with POISSON_SOLVER periodic and PERIODIC xyz.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/ZRAP278MB0827413D164197DF78B9DC76F43F9%40ZRAP278MB0827.CHEP278.PROD.OUTLOOK.COM.
Wiko Ann
Total energy(without SCCS): -17.54128600763402
Total energy(with SCCS): -17.71255174117677
MT
Total energy(without SCCS): -17.49054517094935
-238.953979533671856
-238.946448820809621
-239.402925734069186
-239.260639793435388
-239.180932339281611
-239.014590347465059
-239.163178351021770
-239.194699170551246
-239.162140349715088
Wiko Ann
Krack Matthias (PSI)
Hi
I get the following results with the current CP2K trunk version (epsilon(H2O) = 78.36, cutoff = 1200 Ry)
System Method alpha [mN/m] gamma [mN/m] beta [GPa] rho(min) rho(max) G(Sol) [kcal/mol]
H2O 0d MT 50.0 0.0 -0.350 0.0001 0.0050 -7.16
H2O 3d FFT 50.0 0.0 -0.350 0.0001 0.0050 -7.12
H3O+ 0d MT 5.0 0.0 0.125 0.0002 0.0035 -108.48
OH- 0d MT 0.0 0.0 0.450 0.0024 0.0155 -112.14
using the SCCS parameters from the literature fitted for a plane wave code (Quantum ESPRESSO).
In my opinion, given all the differences between the two code packages CP2K and QE (basis set etc.), these results compare quite well with the experimental values for the free solvation energies
G(Sol, H2O) = -6.3 kcal/mol
G(sol, H3O+) = -108.7 kcal/mol
G(Sol, OH-) = -106.3 kcal/mol
The agreement can be improved, especially for anions, most likely by tuning the SCCS parameters specifically for CP2K.
HTH
.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/4752fc67-7eae-4c54-aef0-c6439b0990b3n%40googlegroups.com.
Krack Matthias (PSI)
Hi Wiko
No clue, never tried combining these features.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/1930068a-0761-4db8-8e79-e10e7c7599f9n%40googlegroups.com.