Wrong density prediction in NpT AIMD
141 views
Skip to first unread message

Abedi, Mostafa
Jul 29, 2020, 8:57:52 AM7/29/20
to cp...@googlegroups.com
Hi Everyone,
I've performed NpT AIMD simulations for a system containing 8 CH4 molecules. The system was equilibrated using 2 ps NVT (300K) followed by 2 ps NVE. For some reason, the predicted density keeps decreasing and getting away from the correct value (The correct density is 0.232 g/cm3):
Time evaluation of density for a box of 8 CH4 at 300 K and 400 bar
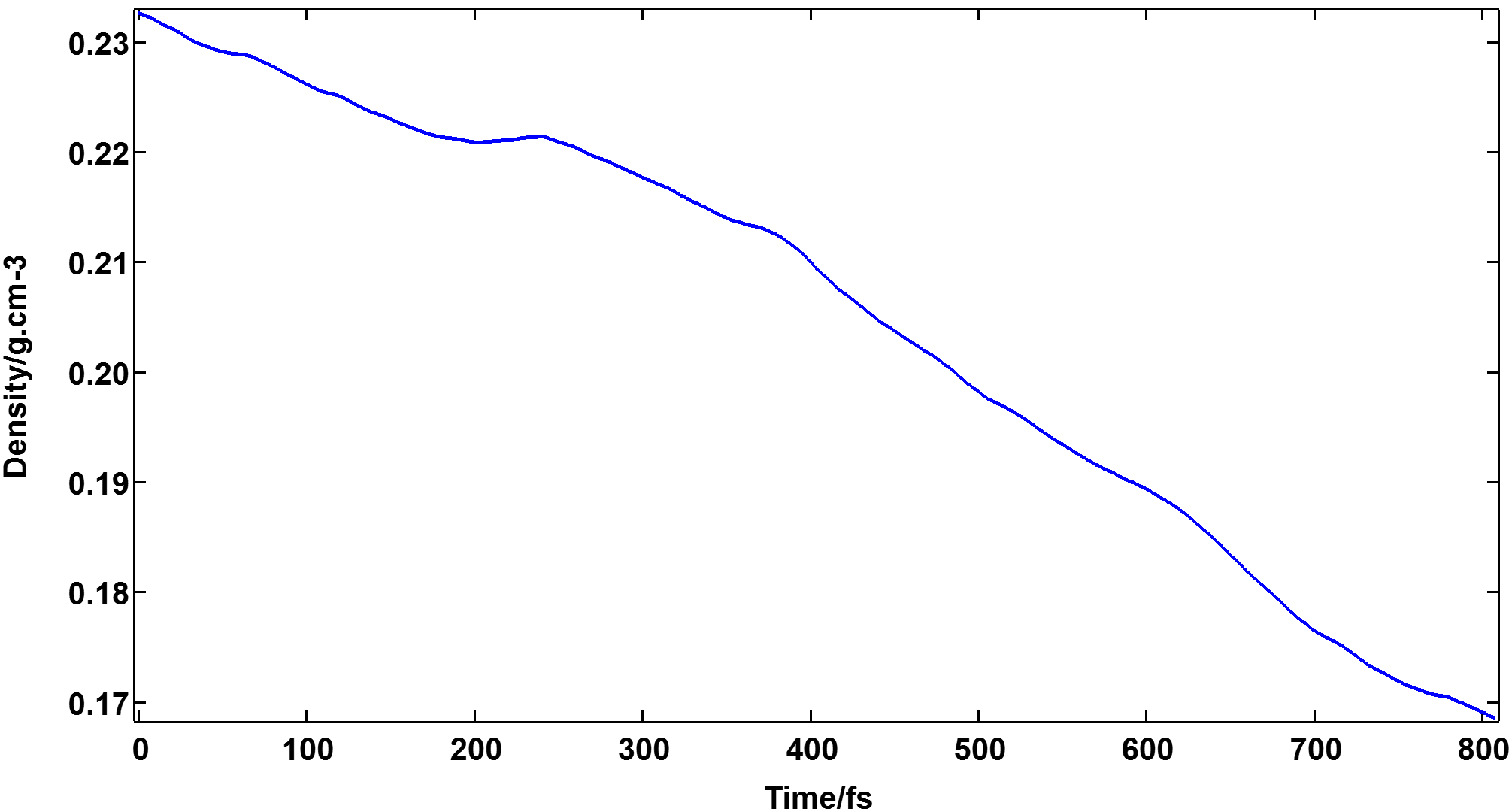
The wB97XD/TZVP-MOLOPT-GTH level of theory was used for the electronic structure part. Unfortunately, there is not much in the literature for this system (or similar systems) to get some ideas for the level of theory. To my mind, it could be due to the wrong prediction of the intermolecular repulsive interactions (vdW dispersion forces). But, there might be some other parameters in the simulations that can cause this issue. Any suggestions or advice for fixing this issue would be greatly appreciated. Many thanks.
Best,
Mostafa
Input file:
&GLOBAL
PROJECT methane
RUN_TYPE MD
WALLTIME 10000000
IOLEVEL LOW
&END GLOBAL
PROJECT methane
RUN_TYPE MD
WALLTIME 10000000
IOLEVEL LOW
&END GLOBAL
&FORCE_EVAL
STRESS_TENSOR ANALYTICAL
METHOD Quickstep
&DFT
BASIS_SET_FILE_NAME BASIS_MOLOPT
POTENTIAL_FILE_NAME GTH_POTENTIALS
CHARGE 0
MULTIPLICITY 1
&MGRID
CUTOFF [Ry] 400
&END
&QS
METHOD GPW
EPS_DEFAULT 1.0E-10
EXTRAPOLATION ASPC
&END
&POISSON
PERIODIC XYZ
POISSON_SOLVER PERIODIC
&END
&SCF
&PRINT
&RESTART OFF
&END
&END
SCF_GUESS ATOMIC
MAX_SCF 300
EPS_SCF 1.0E-6
&OT
PRECONDITIONER FULL_SINGLE_INVERSE
MINIMIZER CG
&END OT
&OUTER_SCF
MAX_SCF 300
EPS_SCF 1.0E-6
&END
&END SCF
&XC
&XC_FUNCTIONAL
&LIBXC
FUNCTIONAL XC_HYB_GGA_XC_WB97X_D
&END LIBXC
&END XC_FUNCTIONAL
&END XC
&END DFT
&SUBSYS
&CELL
ABC [angstrom] 9.71 9.71 9.71
PERIODIC XYZ
&END CELL
&TOPOLOGY
COORD_FILE_NAME methane.xyz
COORD_FILE_FORMAT XYZ
&END
&KIND H
ELEMENT H
BASIS_SET TZVP-MOLOPT-GTH
POTENTIAL GTH-PBE-q1
&END KIND
&KIND C
ELEMENT C
BASIS_SET TZVP-MOLOPT-GTH
POTENTIAL GTH-PBE-q4
&END KIND
&END SUBSYS
&PRINT
&FORCES ON
FILENAME forces
&END
&END
&END FORCE_EVAL
&MOTION
&GEO_OPT
OPTIMIZER BFGS
MAX_ITER 1000
MAX_DR [bohr] 0.003
&BFGS
&END
&END
&MD
ENSEMBLE NPT_I
STEPS 15000
TIMESTEP 0.5
TEMPERATURE 300
&BAROSTAT
PRESSURE 400
&END BAROSTAT
&THERMOSTAT
REGION MASSIVE
TYPE CSVR
&CSVR
TIMECON 50
&END CSVR
&END THERMOSTAT
&END MD
&PRINT
&TRAJECTORY
FORMAT XYZ
&END
&CELL
FILENAME cell
&END
&END
&END MOTION
STRESS_TENSOR ANALYTICAL
METHOD Quickstep
&DFT
BASIS_SET_FILE_NAME BASIS_MOLOPT
POTENTIAL_FILE_NAME GTH_POTENTIALS
CHARGE 0
MULTIPLICITY 1
&MGRID
CUTOFF [Ry] 400
&END
&QS
METHOD GPW
EPS_DEFAULT 1.0E-10
EXTRAPOLATION ASPC
&END
&POISSON
PERIODIC XYZ
POISSON_SOLVER PERIODIC
&END
&SCF
&RESTART OFF
&END
&END
SCF_GUESS ATOMIC
MAX_SCF 300
EPS_SCF 1.0E-6
&OT
PRECONDITIONER FULL_SINGLE_INVERSE
MINIMIZER CG
&END OT
&OUTER_SCF
MAX_SCF 300
EPS_SCF 1.0E-6
&END
&END SCF
&XC
&XC_FUNCTIONAL
&LIBXC
FUNCTIONAL XC_HYB_GGA_XC_WB97X_D
&END LIBXC
&END XC_FUNCTIONAL
&END XC
&END DFT
&SUBSYS
&CELL
ABC [angstrom] 9.71 9.71 9.71
PERIODIC XYZ
&END CELL
&TOPOLOGY
COORD_FILE_NAME methane.xyz
COORD_FILE_FORMAT XYZ
&END
&KIND H
ELEMENT H
BASIS_SET TZVP-MOLOPT-GTH
POTENTIAL GTH-PBE-q1
&END KIND
&KIND C
ELEMENT C
BASIS_SET TZVP-MOLOPT-GTH
POTENTIAL GTH-PBE-q4
&END KIND
&END SUBSYS
&FORCES ON
FILENAME forces
&END
&END
&END FORCE_EVAL
&MOTION
&GEO_OPT
OPTIMIZER BFGS
MAX_ITER 1000
MAX_DR [bohr] 0.003
&BFGS
&END
&END
&MD
ENSEMBLE NPT_I
STEPS 15000
TIMESTEP 0.5
TEMPERATURE 300
&BAROSTAT
PRESSURE 400
&END BAROSTAT
&THERMOSTAT
REGION MASSIVE
TYPE CSVR
&CSVR
TIMECON 50
&END CSVR
&END THERMOSTAT
&END MD
&TRAJECTORY
FORMAT XYZ
&END
&CELL
FILENAME cell
&END
&END
&END MOTION

hut...@chem.uzh.ch
Jul 29, 2020, 10:05:08 AM7/29/20
to cp...@googlegroups.com
Hi
there are a few points to address, some minor, some major:
1) one should use a reference cell (CELL_REF section) in order to
keep the cutoff of the grid consistent.
2) wB97XD is defined using an empirical vdW potential. In your input
this part is missing (it is not in libxc!).
3) You can specify empirical vdW corrections in CP2K in the section
CP2K_INPUT / FORCE_EVAL / DFT / XC / VDW_POTENTIAL / PAIR_POTENTIAL
However, the one used in the definition of wB97XD is not implemented.
The Grimme D2 is close, but uses another damping function, see the original
paper of Chai and Head-Gordon.
regards
Juerg Hutter
--------------------------------------------------------------
Juerg Hutter Phone : ++41 44 635 4491
Institut für Chemie C FAX : ++41 44 635 6838
Universität Zürich E-mail: hut...@chem.uzh.ch
Winterthurerstrasse 190
CH-8057 Zürich, Switzerland
---------------------------------------------------------------
-----cp...@googlegroups.com wrote: -----
To: cp...@googlegroups.com
From: "Abedi, Mostafa"
Sent by: cp...@googlegroups.com
Date: 07/29/2020 02:58PM
Subject: [CP2K:13690] Wrong density prediction in NpT AIMD
Hi Everyone,
I've performed NpT AIMD simulations for a system containing 8 CH4 molecules. The system was equilibrated using 2 ps NVT (300K) followed by 2 ps NVE. For some reason, the predicted density keeps decreasing and getting away from the correct value (The correct density is 0.232 g/cm3):
Time evaluation of density for a box of 8 CH4 at 300 K and 400 bar
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/CAG%3DTvs3qvhzF0fpPAbpQ4GipBmEi-DEq2v80SKbFFjtxMtufPA%40mail.gmail.com.
there are a few points to address, some minor, some major:
1) one should use a reference cell (CELL_REF section) in order to
keep the cutoff of the grid consistent.
2) wB97XD is defined using an empirical vdW potential. In your input
this part is missing (it is not in libxc!).
3) You can specify empirical vdW corrections in CP2K in the section
CP2K_INPUT / FORCE_EVAL / DFT / XC / VDW_POTENTIAL / PAIR_POTENTIAL
However, the one used in the definition of wB97XD is not implemented.
The Grimme D2 is close, but uses another damping function, see the original
paper of Chai and Head-Gordon.
regards
Juerg Hutter
--------------------------------------------------------------
Juerg Hutter Phone : ++41 44 635 4491
Institut für Chemie C FAX : ++41 44 635 6838
Universität Zürich E-mail: hut...@chem.uzh.ch
Winterthurerstrasse 190
CH-8057 Zürich, Switzerland
---------------------------------------------------------------
-----cp...@googlegroups.com wrote: -----
To: cp...@googlegroups.com
From: "Abedi, Mostafa"
Sent by: cp...@googlegroups.com
Date: 07/29/2020 02:58PM
Subject: [CP2K:13690] Wrong density prediction in NpT AIMD
Hi Everyone,
I've performed NpT AIMD simulations for a system containing 8 CH4 molecules. The system was equilibrated using 2 ps NVT (300K) followed by 2 ps NVE. For some reason, the predicted density keeps decreasing and getting away from the correct value (The correct density is 0.232 g/cm3):
Time evaluation of density for a box of 8 CH4 at 300 K and 400 bar
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/CAG%3DTvs3qvhzF0fpPAbpQ4GipBmEi-DEq2v80SKbFFjtxMtufPA%40mail.gmail.com.

Thomas Kühne
Jul 29, 2020, 10:07:45 AM7/29/20
to 'Dorothea Golze' via cp2k
Dear Mostafa,
your simulation is way too short. Equilibrating the density needs many dozens of ps of simulation,
and much more so since you are using the default 1ps time constant for the barostat. My
suggestion is to reduce that to 100 fs and rerun your simulation for at least 25 ps. Also, you were
missing to add the VdW contribution of the wB97X XC functional.
Cheers,
Thomas
Am 29.07.2020 um 14:57 schrieb Abedi, Mostafa <mostaf...@brown.edu>:
Hi Everyone,I've performed NpT AIMD simulations for a system containing 8 CH4 molecules. The system was equilibrated using 2 ps NVT (300K) followed by 2 ps NVE. For some reason, the predicted density keeps decreasing and getting away from the correct value (The correct density is 0.232 g/cm3):Time evaluation of density for a box of 8 CH4 at 300 K and 400 bar
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/CAG%3DTvs3qvhzF0fpPAbpQ4GipBmEi-DEq2v80SKbFFjtxMtufPA%40mail.gmail.com.
==============================
Thomas D. Kühne
Dynamics of Condensed Matter
Chair of Theoretical Chemistry
University of Paderborn
Warburger Str. 100
D-33098 Paderborn
Germany
Abedi, Mostafa
Jul 30, 2020, 1:37:42 PM7/30/20
to cp...@googlegroups.com
Dear
Juerg
and Thomas,
Thank you so much for your helpful suggestions. I will try to modify my input file accordingly and repeat the calculation.
Best,
Mostafa
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/66482C44-5D0D-4C5D-9380-5F31D08F4988%40gmail.com.
Reply all
Reply to author
Forward
0 new messages