Cell Optimization of Platinum (Pt) with fcc (111) surface
284 views
Skip to first unread message

GENG YUAN
Jan 3, 2022, 3:34:56 PM1/3/22
to cp2k
Dear CP2K Users,
Hope you are going great and happy new year.
I am running the cell optimization for a single bulk Pt (134 atoms) with fcc 111 surface. By checking my output file, I notice that the cell optimization takes 446 steps to converge (which is almost close to the maximum number of optimization steps:500). Besides, a certain degree of structure deformation is observed during optimization (please see the attached files and screenshots).
I am wondering if there are any other ways to let the cell optimization converge faster (in other words, converge in fewer steps) and whether the above-mentioned deformation looks reasonable?
Below is the input: (attached are input file, output file, original coordinate, trajectory, and the screenshots of the trajectory)
------------------------------------------------------------
&GLOBAL
PROJECT Pt_cellopt
RUN_TYPE CELL_OPT
PRINT_LEVEL LOW
&END GLOBAL
&FORCE_EVAL
METHOD QS
STRESS_TENSOR ANALYTICAL
&SUBSYS
&CELL
ABC 13.7 13.7 13.7
SYMMETRY CUBIC
&CELL_REF
ABC 13.7*1.5 13.7*1.5 13.7*1.5
&END
&END CELL
&TOPOLOGY
COORD_FILE_NAME ./Pt134.xyz
COORDINATE xyz
&END
&KIND Pt
BASIS_SET DZVP-MOLOPT-SR-GTH
POTENTIAL GTH-PBE-q18
&END KIND
&END SUBSYS
&DFT
BASIS_SET_FILE_NAME ./BASIS_MOLOPT
POTENTIAL_FILE_NAME ./GTH_POTENTIALS
&QS
EPS_DEFAULT 1.0E-12
&END QS
&MGRID
CUTOFF 400
NGRIDS 5
REL_CUTOFF 40
&END MGRID
&SCF
SCF_GUESS ATOMIC
EPS_SCF 1.0E-06
MAX_SCF 200
ADDED_MOS 100
&OUTER_SCF
MAX_SCF 50
EPS_SCF 1.0E-6
&END
&DIAGONALIZATION T
ALGORITHM STANDARD
&END DIAGONALIZATION
&MIXING T
METHOD BROYDEN_MIXING
ALPHA 0.1
NBROYDEN 8
&END MIXING
&SMEAR ON
METHOD FERMI_DIRAC
ELECTRONIC_TEMPERATURE [K] 500
&END SMEAR
&PRINT
&RESTART ON
&END RESTART
&END PRINT
&END SCF
&XC
&XC_FUNCTIONAL
&LIBXC
FUNCTIONAL XC_GGA_X_PBE_R
&END
&LIBXC
FUNCTIONAL XC_GGA_C_PBE
&END
&END XC_FUNCTIONAL
&VDW_POTENTIAL
POTENTIAL_TYPE PAIR_POTENTIAL
&PAIR_POTENTIAL
LONG_RANGE_CORRECTION
PARAMETER_FILE_NAME dftd3.dat
TYPE DFTD3
REFERENCE_FUNCTIONAL revPBE
&END PAIR_POTENTIAL
&END VDW_POTENTIAL
&XC_GRID
XC_DERIV NN50_SMOOTH
&END XC_GRID
&END XC
&END DFT
&END FORCE_EVAL
&MOTION
&CELL_OPT
EXTERNAL_PRESSURE 0
TYPE DIRECT_CELL_OPT
KEEP_SYMMETRY TRUE
MAX_DR 1.0E-05
MAX_FORCE 1.0E-05
RMS_DR 1.0E-05
RMS_FORCE 1.0E-05
MAX_ITER 500
OPTIMIZER BFGS
&END CELL_OPT
&END MOTION
PROJECT Pt_cellopt
RUN_TYPE CELL_OPT
PRINT_LEVEL LOW
&END GLOBAL
&FORCE_EVAL
METHOD QS
STRESS_TENSOR ANALYTICAL
&SUBSYS
&CELL
ABC 13.7 13.7 13.7
SYMMETRY CUBIC
&CELL_REF
ABC 13.7*1.5 13.7*1.5 13.7*1.5
&END
&END CELL
&TOPOLOGY
COORD_FILE_NAME ./Pt134.xyz
COORDINATE xyz
&END
&KIND Pt
BASIS_SET DZVP-MOLOPT-SR-GTH
POTENTIAL GTH-PBE-q18
&END KIND
&END SUBSYS
&DFT
BASIS_SET_FILE_NAME ./BASIS_MOLOPT
POTENTIAL_FILE_NAME ./GTH_POTENTIALS
&QS
EPS_DEFAULT 1.0E-12
&END QS
&MGRID
CUTOFF 400
NGRIDS 5
REL_CUTOFF 40
&END MGRID
&SCF
SCF_GUESS ATOMIC
EPS_SCF 1.0E-06
MAX_SCF 200
ADDED_MOS 100
&OUTER_SCF
MAX_SCF 50
EPS_SCF 1.0E-6
&END
&DIAGONALIZATION T
ALGORITHM STANDARD
&END DIAGONALIZATION
&MIXING T
METHOD BROYDEN_MIXING
ALPHA 0.1
NBROYDEN 8
&END MIXING
&SMEAR ON
METHOD FERMI_DIRAC
ELECTRONIC_TEMPERATURE [K] 500
&END SMEAR
&RESTART ON
&END RESTART
&END PRINT
&END SCF
&XC
&XC_FUNCTIONAL
&LIBXC
FUNCTIONAL XC_GGA_X_PBE_R
&END
&LIBXC
FUNCTIONAL XC_GGA_C_PBE
&END
&END XC_FUNCTIONAL
&VDW_POTENTIAL
POTENTIAL_TYPE PAIR_POTENTIAL
&PAIR_POTENTIAL
LONG_RANGE_CORRECTION
PARAMETER_FILE_NAME dftd3.dat
TYPE DFTD3
REFERENCE_FUNCTIONAL revPBE
&END PAIR_POTENTIAL
&END VDW_POTENTIAL
&XC_GRID
XC_DERIV NN50_SMOOTH
&END XC_GRID
&END XC
&END DFT
&END FORCE_EVAL
&MOTION
&CELL_OPT
EXTERNAL_PRESSURE 0
TYPE DIRECT_CELL_OPT
KEEP_SYMMETRY TRUE
MAX_DR 1.0E-05
MAX_FORCE 1.0E-05
RMS_DR 1.0E-05
RMS_FORCE 1.0E-05
MAX_ITER 500
OPTIMIZER BFGS
&END CELL_OPT
&END MOTION
Many thanks in advance,
Sincerely,
Geng
GENG YUAN
Jan 3, 2022, 3:40:29 PM1/3/22
to cp2k
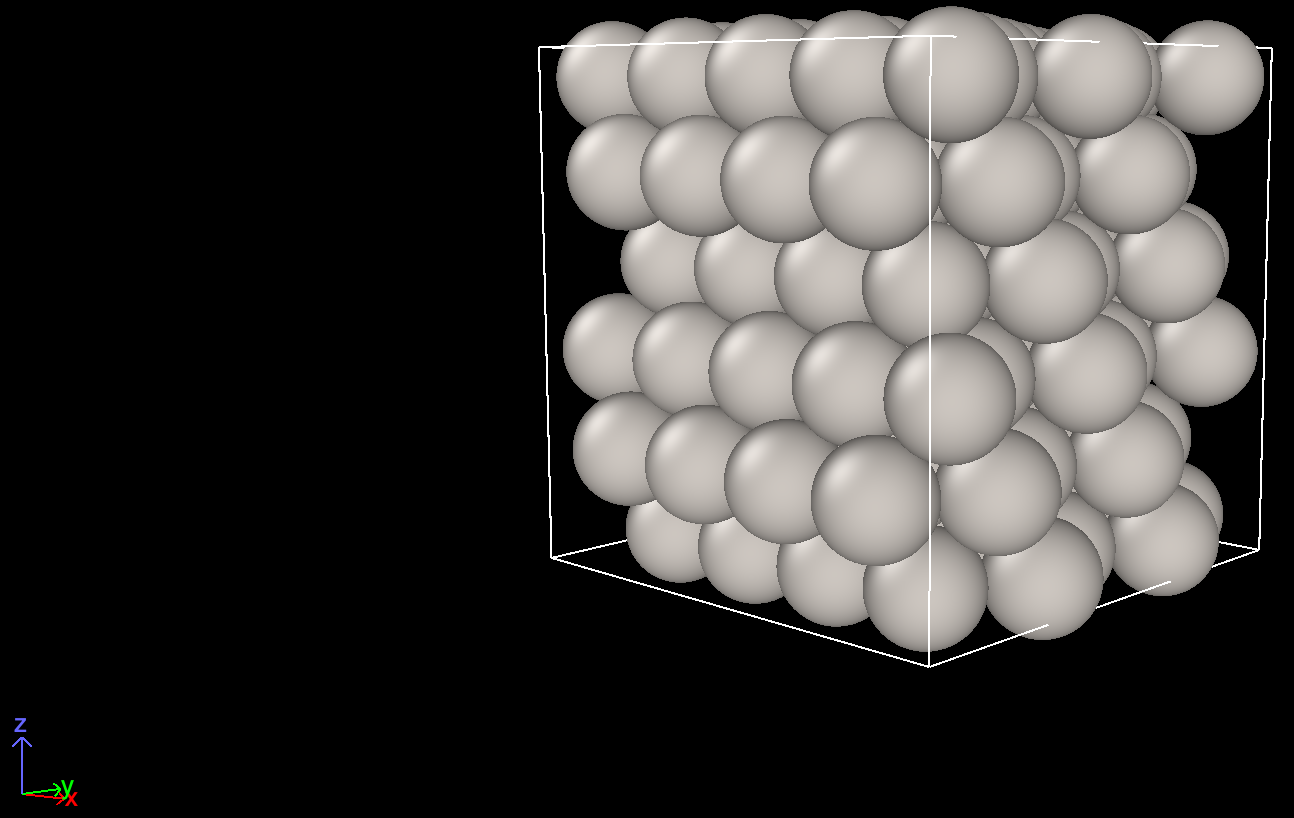
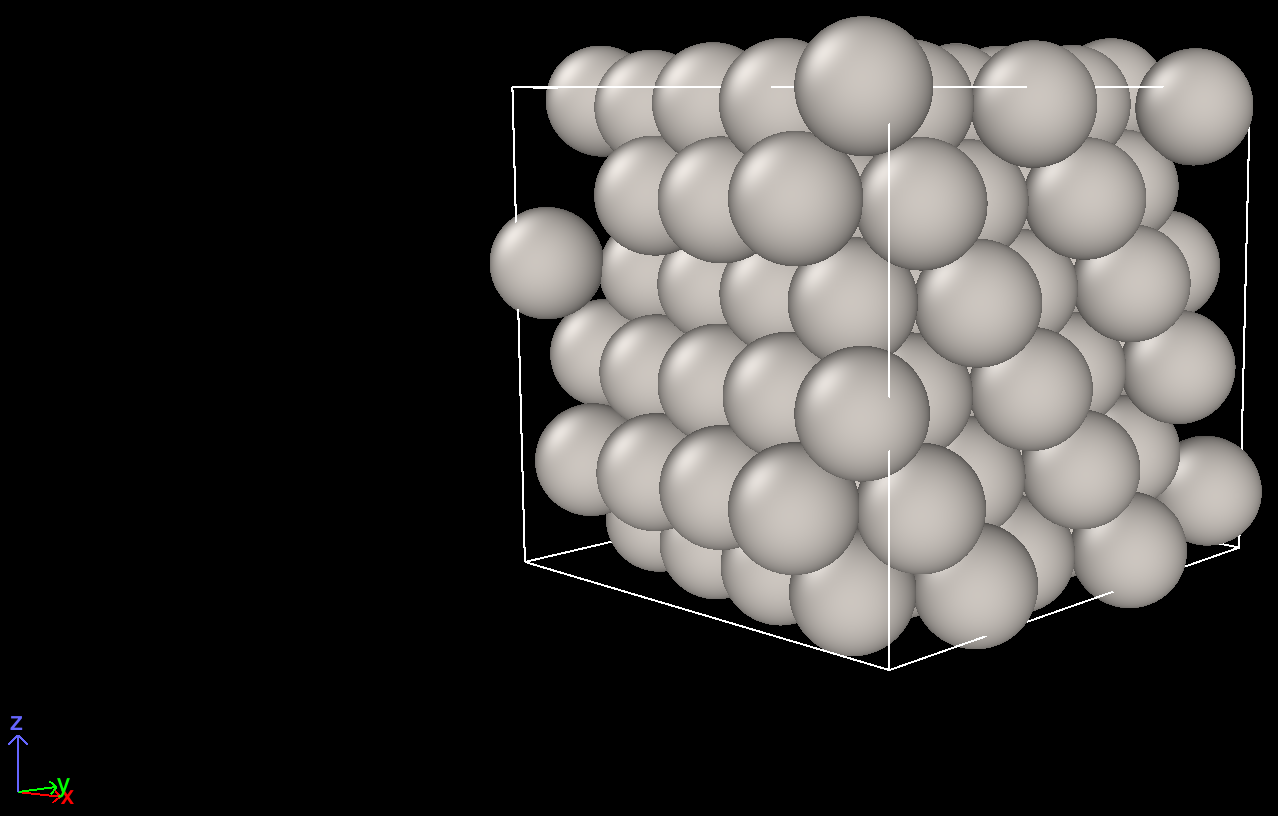
Here are the screenshots.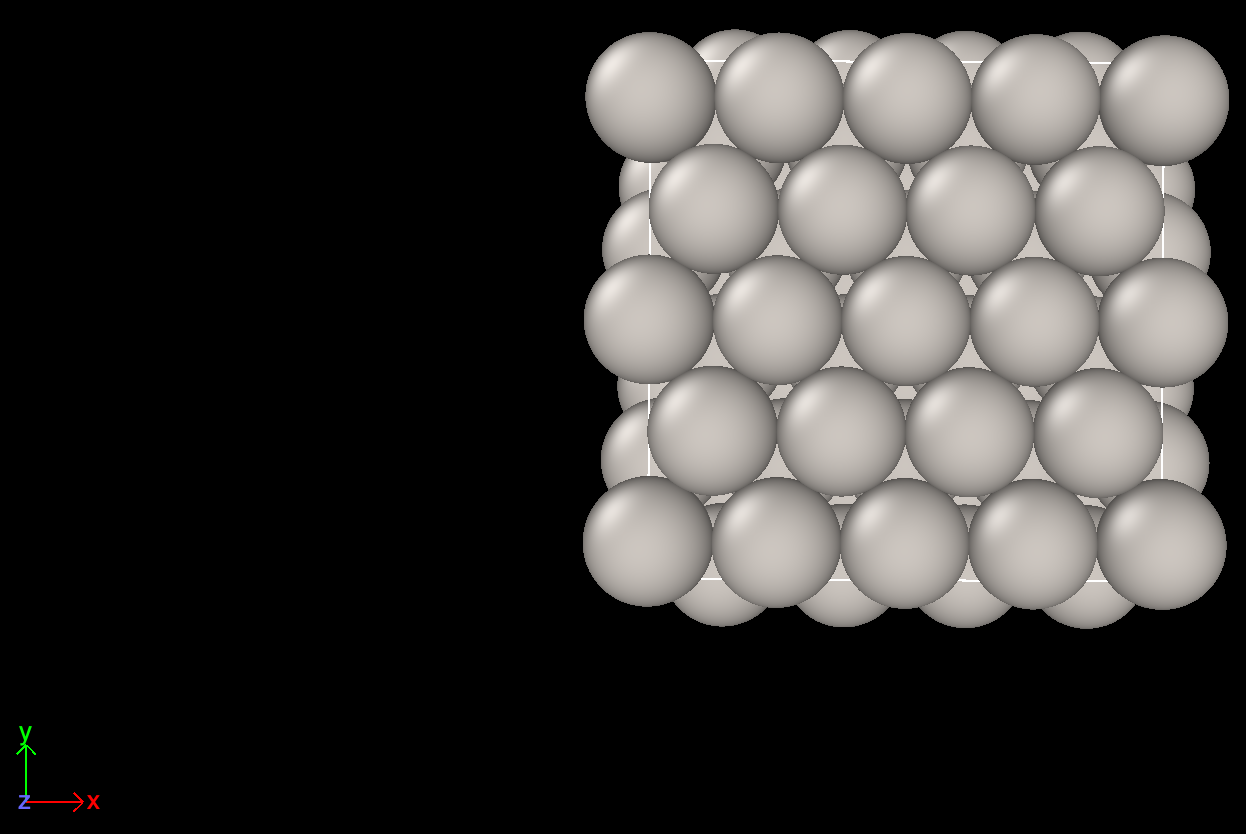
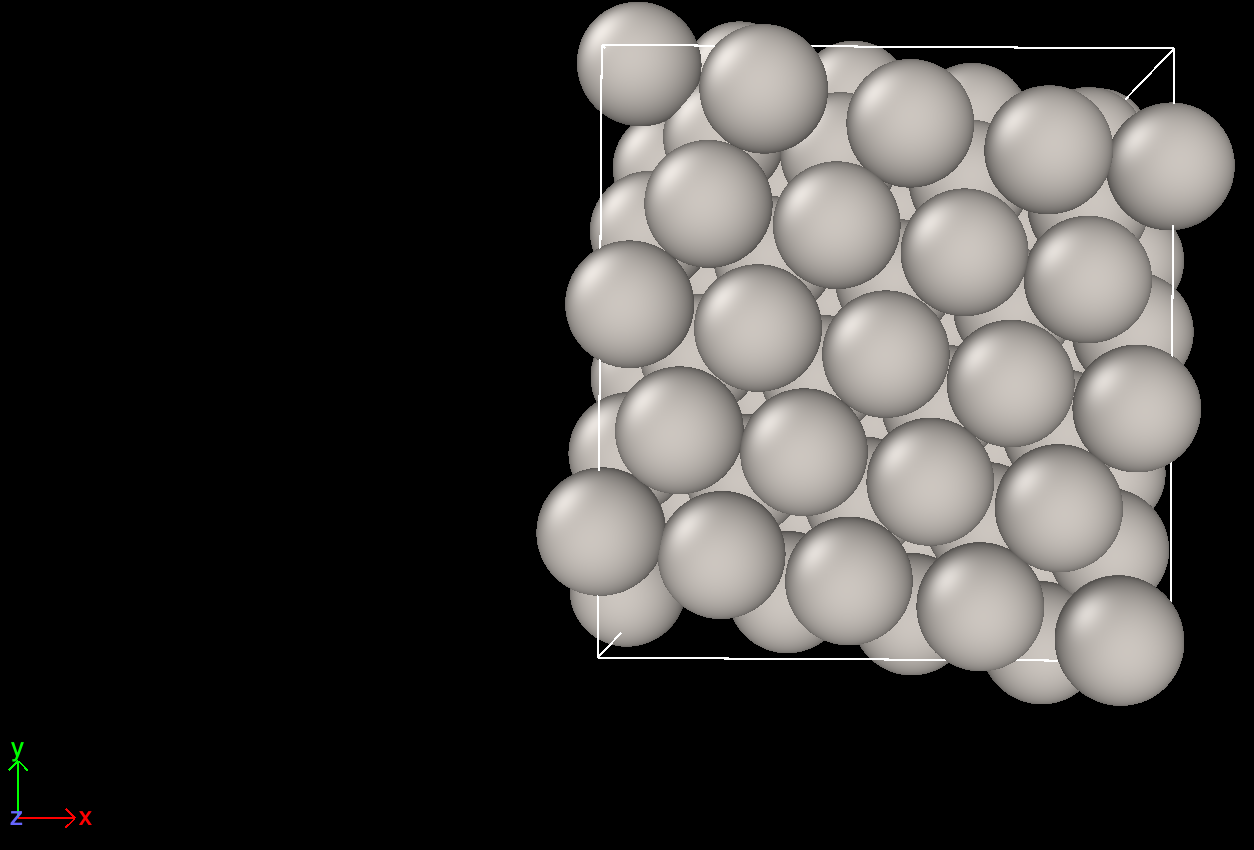
Top view before cell optimization Top view after cell optimization
Another view before cell optimization Another view after cell optimization
Sincerely,
Geng

Fabian Ducry
Jan 3, 2022, 4:03:37 PM1/3/22
to cp...@googlegroups.com
Dear Geng,
You should decrease eps_scf to 1e-7 or 1e-8. The default 1e-6 is good for the initial stage of the optimization, when the forces/pressure are still large. Later on this should be adjusted, especially if you request such tight convergence criteria in &CELL_opt. Decreasing this value reduces the noise in the forces and improves the convergence of the cell optimization.
Cheers,
Fabian
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/d824ebcb-7f45-41ec-9255-25fe50047760n%40googlegroups.com.
GENG YUAN
Jan 5, 2022, 3:49:05 PM1/5/22
to cp2k
Dear Fabian,
Many thanks for your advice!
I decrease eps_scf (under &SCF section) to 1e-8 (the old setting is 1e-6) and found that this didn't improve the convergence of the cell optimization. It took 433 steps to converge, and the overall simulation running time also increased.
Attached are the new input, output, and trajectory for your reference.
Please let me know if any thoughts.
Sincerely,
Geng

Lucas Lodeiro
Jan 5, 2022, 4:00:28 PM1/5/22
to cp...@googlegroups.com
Hi,
I usually set the same EPS_SCF value at the SCF and OUTER_SCF section. Also, I read somewhere (I do not remember), EPS_SCF cannot be less than the square root of EPS_DEFAULT. I guess EPS_DEFAULT 1E-16 and EPS_SCF 1E-8 could converge with less cycles, but the time amount per cycle will be higher.
regards - Lucas
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/239c67b8-664a-4963-a3d1-9100077254d2n%40googlegroups.com.
GENG YUAN
Jan 5, 2022, 4:25:18 PM1/5/22
to cp2k
Dear Lucas,
Many thanks for your information. I didn't notice that the EPS_SCF value under &OUTER_SCF section was still the old value, I will change it the same as the EPS_SCF value under &SCF section.
I will also try to run another test using EPS_DEFAULT 1e-16 to see how the performance looks like.
Sincerely - Geng

lorenzo briccolani
Jan 5, 2022, 4:50:15 PM1/5/22
to cp...@googlegroups.com
Also there is the option EXTRAPOLATION PS under QS section which reads the density directly from the previous scf cycle.
Ps not available for k points
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/c35c3281-90f2-4bbc-aeac-5cfcf48d7a92n%40googlegroups.com.

hut...@chem.uzh.ch
Jan 6, 2022, 5:45:09 AM1/6/22
to cp...@googlegroups.com
Hi
you are asking for extremely tight convergence for the geometry.
10^5 for all criteria is way tighter than the defaults.
If you need this tight convergence, you have to make sure that
gradients are equally accurate. If you check the output, you can see
that the Cell converged way before the geometry.
I would suggest to relax the geometry convergence criteria
or
set
EPS_DEFAULT 10^-14 or lower
EPS_SCF 10^-7 or lower
REL_CUTOFF 60 or higher
CUTOFF 800 or higher
regards
Juerg Hutter
--------------------------------------------------------------
Juerg Hutter Phone : ++41 44 635 4491
Institut für Chemie E-mail: hut...@chem.uzh.ch
Universität Zürich
Winterthurerstrasse 190
CH-8057 Zürich, Switzerland
---------------------------------------------------------------
-----cp...@googlegroups.com wrote: -----
To: "cp2k" <cp...@googlegroups.com>
From: "GENG YUAN"
Sent by: cp...@googlegroups.com
Date: 01/05/2022 10:25PM
Subject: Re: [CP2K:16432] Re: Cell Optimization of Platinum (Pt) with fcc (111) surface
Dear Lucas,
Many thanks for your information. I didn't notice that the EPS_SCF value under &OUTER_SCF section was still the old value, I will change it the same as the EPS_SCF value under &SCF section.
I will also try to run another test using EPS_DEFAULT 1e-16 to see how the performance looks like.
Sincerely - Geng
在2022年1月5日星期三 UTC-5 16:00:28<Lucas Lodeiro> 写道:
Hi,
I usually set the same EPS_SCF value at the SCF and OUTER_SCF section. Also, I read somewhere (I do not remember), EPS_SCF cannot be less than the square root of EPS_DEFAULT. I guess EPS_DEFAULT 1E-16 and EPS_SCF 1E-8 could converge with less cycles, but the time amount per cycle will be higher.
regards - Lucas
El mié, 5 ene 2022 a las 17:49, GENG YUAN (<yuange...@gmail.com>) escribió:
Dear Fabian,
Many thanks for your advice!
I decrease eps_scf (under &SCF section) to 1e-8 (the old setting is 1e-6) and found that this didn't improve the convergence of the cell optimization. It took 433 steps to converge, and the overall simulation running time also increased.
Attached are the new input, output, and trajectory for your reference.
Please let me know if any thoughts.
Sincerely,
Geng
在2022年1月3日星期一 UTC-5 16:03:37<fabia...@gmail.com> 写道:
Dear Geng,
You should decrease eps_scf to 1e-7 or 1e-8. The default 1e-6 is good for the initial stage of the optimization, when the forces/pressure are still large. Later on this should be adjusted, especially if you request such tight convergence criteria in &CELL_opt. Decreasing this value reduces the noise in the forces and improves the convergence of the cell optimization.
Cheers,
Fabian
On 03.01.2022 21:40, GENG YUAN wrote:
Here are the screenshots.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/c35c3281-90f2-4bbc-aeac-5cfcf48d7a92n%40googlegroups.com.
you are asking for extremely tight convergence for the geometry.
10^5 for all criteria is way tighter than the defaults.
If you need this tight convergence, you have to make sure that
gradients are equally accurate. If you check the output, you can see
that the Cell converged way before the geometry.
I would suggest to relax the geometry convergence criteria
or
set
EPS_DEFAULT 10^-14 or lower
EPS_SCF 10^-7 or lower
REL_CUTOFF 60 or higher
CUTOFF 800 or higher
regards
Juerg Hutter
--------------------------------------------------------------
Juerg Hutter Phone : ++41 44 635 4491
Institut für Chemie E-mail: hut...@chem.uzh.ch
Universität Zürich
Winterthurerstrasse 190
CH-8057 Zürich, Switzerland
---------------------------------------------------------------
-----cp...@googlegroups.com wrote: -----
To: "cp2k" <cp...@googlegroups.com>
From: "GENG YUAN"
Sent by: cp...@googlegroups.com
Date: 01/05/2022 10:25PM
Subject: Re: [CP2K:16432] Re: Cell Optimization of Platinum (Pt) with fcc (111) surface
Dear Lucas,
Many thanks for your information. I didn't notice that the EPS_SCF value under &OUTER_SCF section was still the old value, I will change it the same as the EPS_SCF value under &SCF section.
I will also try to run another test using EPS_DEFAULT 1e-16 to see how the performance looks like.
Sincerely - Geng
在2022年1月5日星期三 UTC-5 16:00:28<Lucas Lodeiro> 写道:
Hi,
I usually set the same EPS_SCF value at the SCF and OUTER_SCF section. Also, I read somewhere (I do not remember), EPS_SCF cannot be less than the square root of EPS_DEFAULT. I guess EPS_DEFAULT 1E-16 and EPS_SCF 1E-8 could converge with less cycles, but the time amount per cycle will be higher.
regards - Lucas
El mié, 5 ene 2022 a las 17:49, GENG YUAN (<yuange...@gmail.com>) escribió:
Dear Fabian,
Many thanks for your advice!
I decrease eps_scf (under &SCF section) to 1e-8 (the old setting is 1e-6) and found that this didn't improve the convergence of the cell optimization. It took 433 steps to converge, and the overall simulation running time also increased.
Attached are the new input, output, and trajectory for your reference.
Please let me know if any thoughts.
Sincerely,
Geng
在2022年1月3日星期一 UTC-5 16:03:37<fabia...@gmail.com> 写道:
Dear Geng,
You should decrease eps_scf to 1e-7 or 1e-8. The default 1e-6 is good for the initial stage of the optimization, when the forces/pressure are still large. Later on this should be adjusted, especially if you request such tight convergence criteria in &CELL_opt. Decreasing this value reduces the noise in the forces and improves the convergence of the cell optimization.
Cheers,
Fabian
On 03.01.2022 21:40, GENG YUAN wrote:
Here are the screenshots.
Top view before cell optimization Top view after cell optimization

sumit agrawal
Mar 28, 2022, 2:32:59 AM3/28/22
to cp2k
Hii Geng,
I have little query regarding cell size. Why you consider cell size 13.7 Å. Because from your input lattice structure(pt134.xyz) the cell dimension is r~12.2 Å.
GENG YUAN
Mar 29, 2022, 2:42:09 PM3/29/22
to cp2k
Hi Sumit,
Thanks for your query, actually we used Avogadro Package to build this 111 structure preciously and the cell size is therefore taken from the information shown on the Avogadro package. But later we found that this structure is not what we want and we don't think it is correct/suitable for our simulations. So I would say the cell size in the above input file is not correct.
Many thanks,
Geng
sumit agrawal
Mar 31, 2022, 5:58:24 AM3/31/22
to cp...@googlegroups.com
Hi Geng,
Thanks for your gentle reply. One more query.
Can you tell me how to choose a simulation box size (In &CELL of input file)? Suppose if my lattice dimension for crystal a=b=c is 5 Å. And if I use the same length for my simulation box (In section &CELL of my input file) , then one error appears ("atoms are too close"). Then I increase the simulation box length to 6 or 7 Å (in my &CELL section), then this error is disappear. Here I am using PBC for my simulation.
So can you comment on this observation? How far can we go from lattice dimension for selecting the simulation box size?
Thanks
sumit
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/89a8ca45-20bc-465b-81b0-feac50b96f33n%40googlegroups.com.
GENG YUAN
Apr 1, 2022, 10:56:20 AM4/1/22
to cp2k
Hi Sumit,
As far as I know, it is because you use PBC, so the inappropriate size of the simulation box may bring some atoms too close when this box is duplicated in all directions (Assume PBC in all directions).
Could you share one example of your system (e.g. XYZ file may be perfect) so we can have a look?
Sincerely,
Geng
Reply all
Reply to author
Forward
0 new messages