IR spectrum. Help wanted.

z sh
Hello everyone,
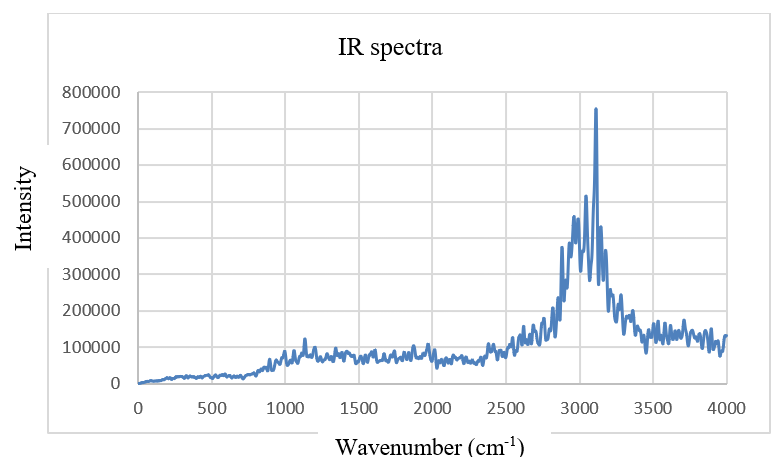
I am trying to do vibrational analysis for gas phase system. I am following the tutorial "Calculating Vibrational Spectra from Molecular Dynamics". Initially i have done massive equilibration of a single methanol system for 6000 steps. After that i use the following example CP2k input file to simulating a trajectory with wannier centers for 60000 steps. Then, I calculated the IR spectra with methanol_wannier.xyz file using Travis software. Unfortunately, I don’t get a good IR spectrum because of it doesn’t show peaks at 200, 1000, 1500 and 3700 cm-1 and it has very noise. Would you please guide me why I don’t get a good IR spectrum?
Here i am attaching my IR spectrum and the input files of massive equilibration and the wannier center for a single methanol.
Please help me on this.
Thanks.

Sam Broderick
Sam Broderick

Thomas Kühne
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/65704ff7-7bd2-4854-886c-074a23c4f9a0n%40googlegroups.com.
Sam Broderick
apply for Raman and ROA spectra. The intensity is specified in atomic units. In the tutorial,
we use a value of 5 · 10 −3 a.u. = 2.57 · 10 9 V m −1 , which we found to be a good compromise.
Too small values lead to noisy spectra, whereas too large values might leave the linear regime
of electronic polarizability."