CELL Optimization - Energy not decreasing
389 views
Skip to first unread message

Jan Elsner
Apr 18, 2020, 2:33:24 PM4/18/20
to cp...@googlegroups.com
Dear all,
I am trying to run a Cell Optimisation on a periodic molecular crystal (beta-resorcinol: C6H6O2, 4 molecules in unit cell) using K-points and BFGS as optimiser. I am using the vdW-DF-optPBE functional (https://github.com/cp2k/cp2k/commit/74ef05b96a46523a0daf7da04e75804420b5cd5e). After 83 optimisation steps, my system does converge to the required MOTION thresholds, however from step 7 onwards the total energy increases at every step i.e.:
-------- Informations at step = 7 ------------
Optimization Method = BFGS
Total Energy = -280.1489665596
Internal Pressure [bar] = -2667.4573178334
Real energy change = 0.0001037473
Predicted change in energy = -0.0001063956
Scaling factor = 0.0000000000
Step size = 0.0124910496
Trust radius = 0.4724315332
Decrease in energy = NO
Used time = 60.978
Convergence check :
Max. step size = 0.0124910496
Conv. limit for step size = 0.0010000000
Convergence in step size = NO
RMS step size = 0.0029192108
Conv. limit for RMS step = 0.0010000000
Convergence in RMS step = NO
Max. gradient = 0.0034738629
Conv. limit for gradients = 0.0001000000
Conv. for gradients = NO
RMS gradient = 0.0006857738
Conv. limit for RMS grad. = 0.0001000000
Conv. for gradients = NO
Pressure Deviation [bar] = -2668.4705678334
Pressure Tolerance [bar] = 200.0000000000
Conv. for PRESSURE = NO
---------------------------------------------------
The resulting cell-optimised structure is therefore not a minimum in energy. I've attached the input and output files. Some points which may (or may not) be relevant:
- I did not encounter this problem when using a different functional (PBE + D3 instead of optPBE-vdW as used here), but otherwise identical settings.
- I encountered the same issue (energy increasing at every step) using CUTOFF/REL_CUTOFF = 800/60
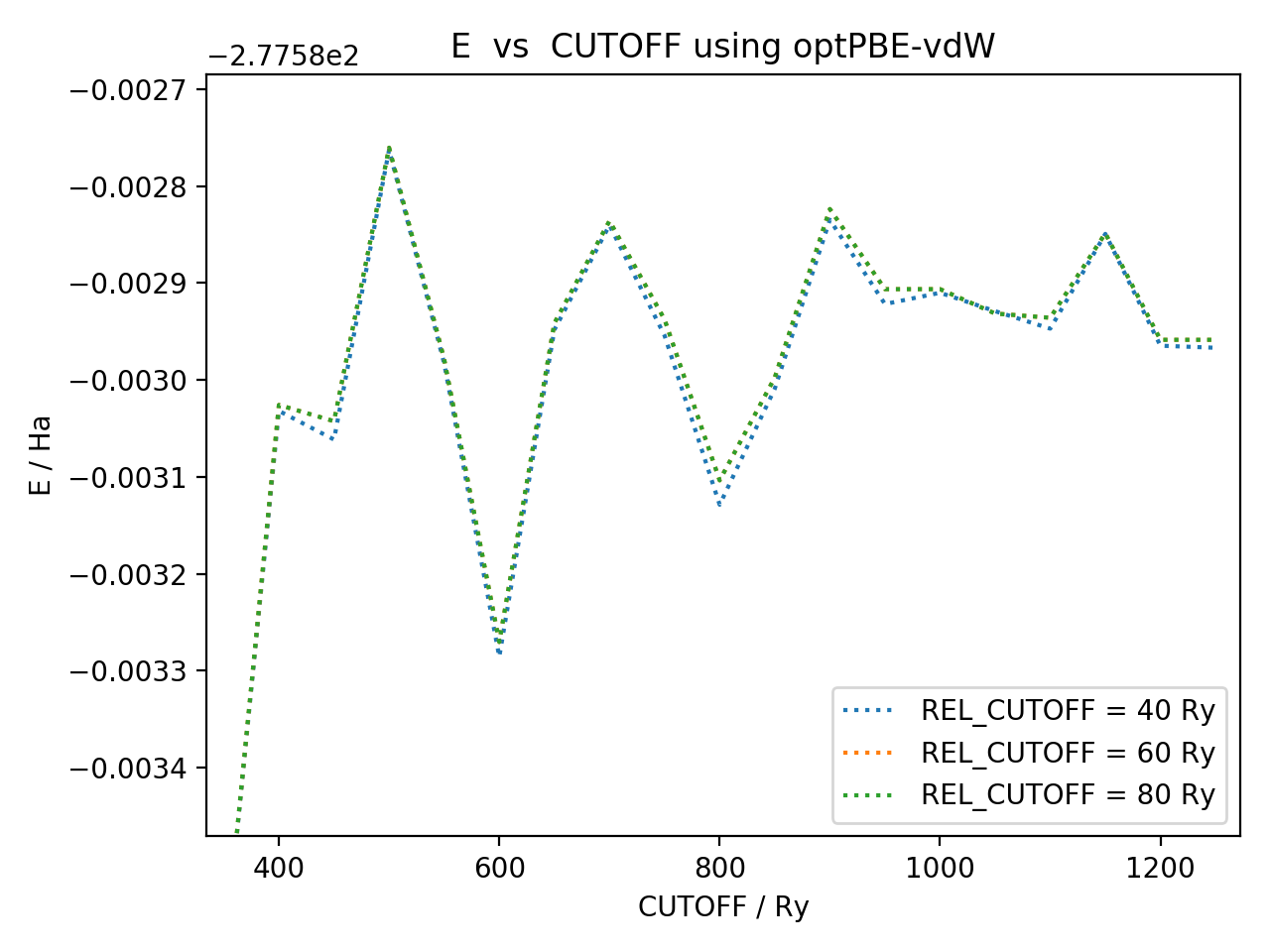
- I initially tried to converge CUTOFF and REL_CUTOFF as in the tutorial (https://www.cp2k.org/howto:converging_cutoff), however my single point calculations did not converge quite as nicely as in the tutorial - instead oscillating with an amplitude of order ~ e-4 Ha (I've attached a figure showing this). This is why I use such large CUTOFF/REL_CUTOFF values. I note that this was also the case when I ran PBE + D3 calculations, however in the latter case my cell optimisation did work.
Any input would be greatly appreciated.
Best wishes,
Jan

hut...@chem.uzh.ch
Apr 20, 2020, 5:20:34 AM4/20/20
to cp...@googlegroups.com
Hi
I don't know how well a setup as this is tested. However, from
your output I would guess the problem could be related to your
SCF convergence. With the methods you are using you have to set
the convergence criteria much tighter in order to get converged
energy and forces. I would suggest to use at least 1.E-8.
The ultimate test would be to do a DEBUG run in order to verify
forces and stress tensor for the setting of options.
regards
Juerg Hutter
--------------------------------------------------------------
Juerg Hutter Phone : ++41 44 635 4491
Institut für Chemie C FAX : ++41 44 635 6838
Universität Zürich E-mail: hut...@chem.uzh.ch
Winterthurerstrasse 190
CH-8057 Zürich, Switzerland
---------------------------------------------------------------
-----cp...@googlegroups.com wrote: -----
To: "cp2k" <cp...@googlegroups.com>
From: "Jan Elsner"
Sent by: cp...@googlegroups.com
Date: 04/18/2020 08:33PM
Subject: [CP2K:13145] CELL Optimization - Energy not decreasing
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/e97788c6-1c43-453d-893e-6b973a977f9d%40googlegroups.com.
[attachment "cell.inp" removed by Jürg Hutter/at/UZH]
[attachment "cell.out" removed by Jürg Hutter/at/UZH]
[attachment "E_vs_cutoff.png" removed by Jürg Hutter/at/UZH]
[attachment "subsys.include" removed by Jürg Hutter/at/UZH]
I don't know how well a setup as this is tested. However, from
your output I would guess the problem could be related to your
SCF convergence. With the methods you are using you have to set
the convergence criteria much tighter in order to get converged
energy and forces. I would suggest to use at least 1.E-8.
The ultimate test would be to do a DEBUG run in order to verify
forces and stress tensor for the setting of options.
regards
Juerg Hutter
--------------------------------------------------------------
Juerg Hutter Phone : ++41 44 635 4491
Institut für Chemie C FAX : ++41 44 635 6838
Universität Zürich E-mail: hut...@chem.uzh.ch
Winterthurerstrasse 190
CH-8057 Zürich, Switzerland
---------------------------------------------------------------
-----cp...@googlegroups.com wrote: -----
To: "cp2k" <cp...@googlegroups.com>
From: "Jan Elsner"
Sent by: cp...@googlegroups.com
Date: 04/18/2020 08:33PM
Subject: [CP2K:13145] CELL Optimization - Energy not decreasing
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/e97788c6-1c43-453d-893e-6b973a977f9d%40googlegroups.com.
[attachment "cell.inp" removed by Jürg Hutter/at/UZH]
[attachment "cell.out" removed by Jürg Hutter/at/UZH]
[attachment "E_vs_cutoff.png" removed by Jürg Hutter/at/UZH]
[attachment "subsys.include" removed by Jürg Hutter/at/UZH]
Jan Elsner
Apr 24, 2020, 5:43:00 AM4/24/20
to cp2k
Dear Juerg,
Many thanks for the quick response! Unfortunately, decreasing EPS_SCF (all the way to 1.0E-10) does not solve the problem.
I have done a DEBUG run which results in the following error (see end of attached file debug.out): 'Abort: A mismatch between the analytical and the numerical stress tensor has been detected. Check the implementation of the stress tensor'. I'm not entirely sure how I should proceed from here / what the problem is exactly - any insight would be very much appreciated.
If it helps shed some light on the problem, I also attach a figure showing Total Energy, RMS Gradient and Cell Volume as a function of optimization step (plotted from the original cell.out file I sent). The optimiser seems to overshoot the minimum energy configuration.
One other question which comes to mind is whether it's ok to use GTH-PBE or GTH-PADE pseudopotentials with optPBE-vdW?
Best wishes,
Jan
Jan
To unsubscribe from this group and stop receiving emails from it, send an email to cp...@googlegroups.com.
Jan Elsner
Apr 30, 2020, 11:30:49 AM4/30/20
to cp2k
Dear all,
I thought I should post an update on this.
It seems that there is a problem with using PW92 as the local correlation part of XC functional. When I use VWN instead, the analytical and numerical forces are equal. I attach input and output files for DEBUG runs on a single H20 molecule using optPBE in combination with PW92 and VWN. In the case of PW92, there is a mismatch between analytical and numerical forces.
I note that the same applies for the some of the other functionals listed here: https://github.com/cp2k/cp2k/tree/master/data/xc_section (at least, I can confirm that optB88 and C09 also give a mismatch between numerical and analytical forces using the settings listed here).
Best wishes,
Jan

Thomas Kühne
Apr 30, 2020, 12:14:33 PM4/30/20
to cp...@googlegroups.com
Dear Jan,
thanks for putting this to our attention, the deviation looks indeed on the upper end.
However, computing numerical derivatives is a bit tricky and has to be done with
maximum accuracy, since relative deviations of small values are reported.
So this may be an error, but can you please increase EPS_DEFAULT to 10E-16
and EPS_SCF to 10E-7 and try this also with DX to 5.0E-4?
Cheers,
Thomas
P.S. BTW, I did a similar test with the sole PADE XC functional, which is supposed
to mimic PW92 LDA many years ago and it passed the test ...
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/48cb2870-f660-437b-83b2-4a9e54e15b20%40googlegroups.com.
<pw92.inp><pw92.out><vwn.inp><vwn.out>
==============================
Thomas D. Kühne
Dynamics of Condensed Matter
Chair of Theoretical Chemistry
University of Paderborn
Warburger Str. 100
D-33098 Paderborn
Germany
hut...@chem.uzh.ch
Apr 30, 2020, 12:24:26 PM4/30/20
to cp...@googlegroups.com
Hi
yes, I cam to the same conclusion. The problem seems to be
the value for rs where the different formulas have to be switched.
Changing at line 117 in xc_perdew_wang.fypp the value from
1.0 to 0.5 seems to solve most problems.
In addition, you might also want to use a reference cell (see CELL_REF)
when optimizing cells.
I will update the Trunk version of CP2K soon.
regards
Juerg Hutter
--------------------------------------------------------------
Juerg Hutter Phone : ++41 44 635 4491
Institut für Chemie C FAX : ++41 44 635 6838
Universität Zürich E-mail: hut...@chem.uzh.ch
Winterthurerstrasse 190
CH-8057 Zürich, Switzerland
---------------------------------------------------------------
-----cp...@googlegroups.com wrote: -----
To: "cp2k" <cp...@googlegroups.com>
From: "Jan Elsner"
Sent by: cp...@googlegroups.com
Date: 04/30/2020 05:31PM
Subject: Re: [CP2K:13207] CELL Optimization - Energy not decreasing
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/48cb2870-f660-437b-83b2-4a9e54e15b20%40googlegroups.com.
[attachment "pw92.inp" removed by Jürg Hutter/at/UZH]
[attachment "pw92.out" removed by Jürg Hutter/at/UZH]
[attachment "vwn.inp" removed by Jürg Hutter/at/UZH]
[attachment "vwn.out" removed by Jürg Hutter/at/UZH]
yes, I cam to the same conclusion. The problem seems to be
the value for rs where the different formulas have to be switched.
Changing at line 117 in xc_perdew_wang.fypp the value from
1.0 to 0.5 seems to solve most problems.
In addition, you might also want to use a reference cell (see CELL_REF)
when optimizing cells.
I will update the Trunk version of CP2K soon.
regards
Juerg Hutter
--------------------------------------------------------------
Juerg Hutter Phone : ++41 44 635 4491
Institut für Chemie C FAX : ++41 44 635 6838
Universität Zürich E-mail: hut...@chem.uzh.ch
Winterthurerstrasse 190
CH-8057 Zürich, Switzerland
---------------------------------------------------------------
-----cp...@googlegroups.com wrote: -----
To: "cp2k" <cp...@googlegroups.com>
From: "Jan Elsner"
Sent by: cp...@googlegroups.com
Subject: Re: [CP2K:13207] CELL Optimization - Energy not decreasing
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/48cb2870-f660-437b-83b2-4a9e54e15b20%40googlegroups.com.
[attachment "pw92.inp" removed by Jürg Hutter/at/UZH]
[attachment "pw92.out" removed by Jürg Hutter/at/UZH]
[attachment "vwn.inp" removed by Jürg Hutter/at/UZH]
[attachment "vwn.out" removed by Jürg Hutter/at/UZH]
Jan Elsner
Apr 30, 2020, 2:04:56 PM4/30/20
to cp2k
Dear Thomas,
I attach inputs and outputs with the modified settings. As before, using VWN gives consistent forces. Using PW92 gives inconsistent forces (as before) however now the forces for Atom 2 are fine, but way off for Atom 3. I'm not sure why this would be the case.
Best,
Jan
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/48cb2870-f660-437b-83b2-4a9e54e15b20%40googlegroups.com.
<pw92.inp><pw92.out><vwn.inp><vwn.out>
Jan Elsner
Apr 30, 2020, 2:06:06 PM4/30/20
to cp2k
Dear Juerg,
Ok, cheers for the advice.
Best wishes,
Jan

{kind=link}
{kind=link}
Krack Matthias (PSI)
Apr 30, 2020, 2:19:22 PM4/30/20
to cp...@googlegroups.com
Dear Jan
You are right, the PW92 has been buggy and gave different numbers compared to the libxc version XC_LDA_C_PW which you could use alternatively. After Jürg’s fix for PW92, the stress tensor for your system looks fine now within the numerical tolerances, though the debug run might still end with an error message, since the default test criterion of 1.0E-5 for sum of the deviations for all tensor elements is quite tight.
Best
Matthias
To unsubscribe from this group and stop receiving emails from it, send an email to
cp2k+uns...@googlegroups.com.
To view this discussion on the web visit
https://groups.google.com/d/msgid/cp2k/b588cc61-36a0-4fe8-a4fa-f97f46625603%40googlegroups.com.
Reply all
Reply to author
Forward
0 new messages