MOF cell optimization
93 views
Skip to first unread message

Lei Chen
Jul 13, 2022, 3:01:02 AM7/13/22
to cp2k
Hi there,
I’m doing the cell
optimization of phthalocyanine-based MOF structure and I found that result is
quite different from the literature. Here is the result I got and the one in
some paper.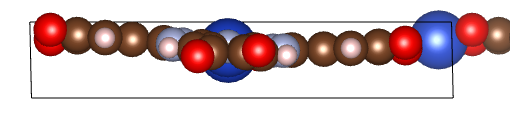

It looks that the one I optimized using cp2k shows some twist and is not perfect planar structure. I think it should be the problem of parameter setting. But I get similar structure after increasing the convergence criterion and changing optimizer. So I want to check if there is any other prime parameter that affect the structure optimization result? here is the parameter in the paper using vasp and my input file of cp2k is attached:

Thanks for your help and any suggestion.
Sincerely
Lei Chen

Quentin Pessemesse
Jul 13, 2022, 3:49:45 AM7/13/22
to cp...@googlegroups.com
Hello,
I am not a VASP expert but from your input files, there are differences between the reported level of theory and the one you are using.
1. BASIS_SET DZVP-MOLOPT-SR-GTH
You are using a gaussian-plane-wave (GPW) scheme with the MOLOPT basis set and plane waves with a cutoff at 400 Ry while the authors report using a plane-wave only scheme with a cutoff at 500 Ry.
2. SCHEME MONKHORST-PACK 1 1 6
You are sampling the Brillouin zone with a 1 x 1 x 6 grid, while the authors report using a 4 x 4 x 1 grid. I believe this can make a large difference as you are sampling the Brillouin zone very differently.
3. PARAMETER_FILE_NAME dftd3.dat
You are using the D3 dispersion correction while authors use the D2 correction.
4. METHOD FERMI_DIRAC
You are using Fermi smearing which the authors do not report using.
5. MULTIPLICITY 2
You are setting the spin multiplicity in your calculations while I believe the spin multiplicity is relaxed by default in VASP spin-polarized calculations. You set a multiplicity of 2 which may not be the ground state of your system, especially if more than 1 Cu atom is Cu(II).
I would say points 2 & 5 are most likely responsible for your issue, but maybe someone with more expertise in VASP can weigh in.
Take care,
Quentin PESSEMESSE
Univ Lyon / ETH Zurich
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/270ae033-2f61-47aa-9c46-f5eaf1b75069n%40googlegroups.com.
Reply all
Reply to author
Forward
0 new messages