Error with "Cholesky decompose failed"
314 views
Skip to first unread message

Jiapeng Liu
Sep 30, 2019, 12:38:35 PM9/30/19
to cp2k
Dear CP2K users,
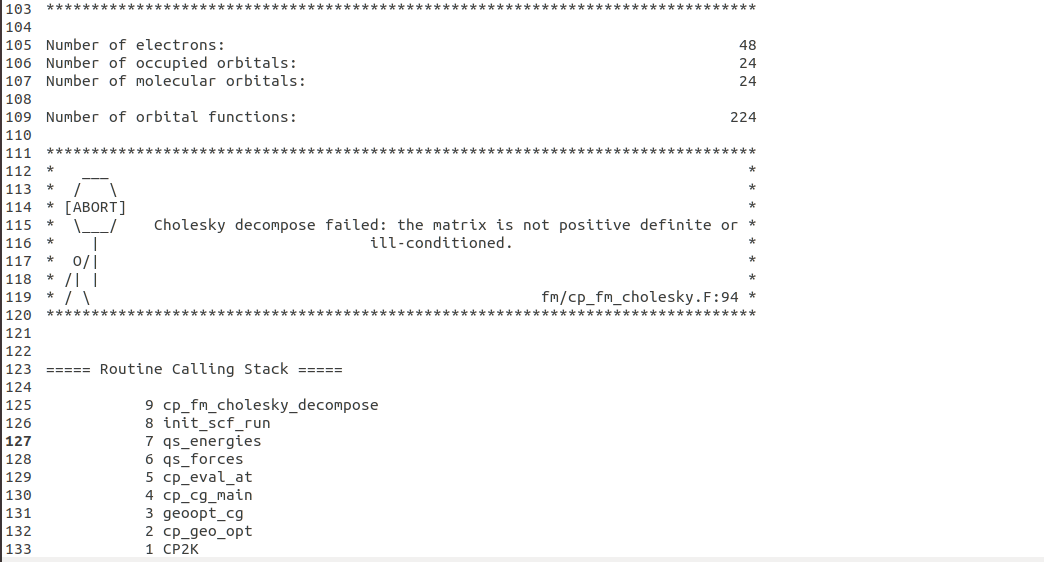
I am just learning CP2K from the official website and trying to relax a Li bcc structure where 16 atoms are included. I modified from the example shown here https://www.cp2k.org/exercises:2019_conexs_newcastle:ex3#part_1optimizing_geometry for MgO. But I met some problems running my input file. The output says that "Cholesky decompose failed: the matrix is not positive definite or ill-conditioned". I am quite confused where is the error from. Can you guys help me on this. Thanks for your kind help.
I also attach my input file here, thank you very much.

Patrick Gono
Oct 1, 2019, 5:37:37 AM10/1/19
to cp...@googlegroups.com
Dear Jiapeng,
I managed to avoid this error by decreasing the value of EPS_PGF_ORB to 1E-10 from the default value of SQRT(EPS_DEFAULT) = 1E-5:
&QS
METHOD GPW ! to optimize the geometry the GPW method will be used
EPS_PGF_ORB 1E-10
&END QS
METHOD GPW ! to optimize the geometry the GPW method will be used
EPS_PGF_ORB 1E-10
&END QS
However, while the calculation runs, the SCF loop does not converge. I suggest you use an orbital transformation method (section &OT). Unless you need to deal with extra orbitals, smearing, or other methods that clash with the use of OT, you should treat is as the preferred option. Likewise, using an inner and an outer SCF loop will make the calculation more robust and converge faster:
&SCF
MAX_SCF 100
EPS_SCF 1.0E-6
SCF_GUESS ATOMIC
&OT
MINIMIZER CG
PRECONDITIONER FULL_KINETIC
&END OT
&OUTER_SCF
EPS_SCF 1.0E-6
MAX_SCF 10
&END OUTER_SCF
&END SCF
MAX_SCF 100
EPS_SCF 1.0E-6
SCF_GUESS ATOMIC
&OT
MINIMIZER CG
PRECONDITIONER FULL_KINETIC
&END OT
&OUTER_SCF
EPS_SCF 1.0E-6
MAX_SCF 10
&END OUTER_SCF
&END SCF
I attach the modified input to this email for the sake of convenience.
Yours sincerely,
Patrick Gono
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/d849b103-df14-456a-8dee-3556d26d5156%40googlegroups.com.
Jiapeng Liu
Oct 1, 2019, 8:10:30 AM10/1/19
to cp2k
Dear Patrick Gono,
Thanks for your kind help and very useful suggestions. I tried with your script and the problem seems solved. I am just a beginner of CP2K and found that the SCF does not converge for 100 steps. I am still trying to tune some parameters on it. Thanks.
Regards,
Jiapeng
To unsubscribe from this group and stop receiving emails from it, send an email to cp...@googlegroups.com.
Message has been deleted
Patrick Gono
Oct 1, 2019, 11:55:40 AM10/1/19
to cp...@googlegroups.com
Dear Jiapeng
Following the reply by Travis, I must apologize -- I did not realize pure Lithium is a metal (rare earth metals, duh!). Needless to say, please follow the advice of Travis and adopt smearing.
Yours sincerely,
Patrick Gono
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/1945e707-b119-4a68-aa37-e57c97a82063%40googlegroups.com.
Jiapeng Liu
Oct 1, 2019, 10:07:20 PM10/1/19
to cp2k
Hi Travis,
Really thanks for your good suggestion. I will try this set of parameters and let you know if there are some problems. Thanks.
Best,
Jiapeng
On Tuesday, October 1, 2019 at 10:50:17 PM UTC+8, Travis wrote:
On Tuesday, October 1, 2019 at 10:50:17 PM UTC+8, Travis wrote:
Hi,Metallic systems need Fermi-Dirac smearing to converge. Something like this is more fitting. Add in a section for GEO_OPT or CELL_OPT and add coordinates and a KIND section. The bits in bold are most pertinent to your case.
METHOD QUICKSTEP
STRESS_TENSOR ANALYTICAL
&DFT
UKS T
CHARGE 0
MULTIPLICITY 1
BASIS_SET_FILE_NAME data/BASIS_MOLOPT
POTENTIAL_FILE_NAME data/POTENTIAL
&MGRID
CUTOFF 800
NGRIDS 5
RELATIVE_CUTOFF 50
&END MGRID
&QS
EPS_DEFAULT 1.0E-12
METHOD GPW
EXTRAPOLATION USE_GUESS
&END QS
&SCF
EPS_SCF 1e-06
MAX_SCF 200
SCF_GUESS RESTART
ADDED_MOS 400
&SMEAR T
METHOD FERMI_DIRAC
ELECTRONIC_TEMPERATURE 3.0000000000000000E+02
&END SMEAR
&MIXING T
METHOD BROYDEN_MIXING
ALPHA 4.0000000000000002E-01
NMIXING 5
NBUFFER 8
&END MIXING
&END SCF
&XC
FUNCTIONAL_ROUTINE NEW
DENSITY_CUTOFF 1.0e-12
GRADIENT_CUTOFF 1.0e-12
TAU_CUTOFF 1.0e-12
&XC_FUNCTIONAL
&PBE
PARAMETRIZATION Orig
&END PBE
&END XC_FUNCTIONAL
&XC_GRID
USE_FINER_GRID T
&END XC_GRID
&END XC
&POISSON
POISSON_SOLVER PERIODIC
PERIODIC XYZ
&END POISSON
&KPOINTS
SCHEME MONKHORST-PACK 4 4 4
FULL_GRID .TRUE.
&END KPOINTS
&END DFT
&SUBSYS
&CELL
ABC 6.7638 6.7638 6.7638
ALPHA_BETA_GAMMA 90.000000 90.000000 90.000000
PERIODIC XYZ
MULTIPLE_UNIT_CELL 1 1 1
&END CELL
...
&END SUBSYS
&END FORCE_EVALAdditionally, note that the Cholesky decomposition error is usually related to using too large a basis set or too diffuse a basis set for the problem. For a double-zeta flavor of basis set, the EPS_PGF_ORB fix is a reasonable approach to correct the issue. But for triple-zeta and quadruple-zeta (or higher) basis sets, it is usually better to select a smaller or less diffuse basis set. It is very common to use the MOLOPT-SR variants as they do well for solids and condensed phase simulations. The short-ranged (SR) variant is purpose built to be less diffuse, so it's more likely any overlap issues come from poor initial geometry, even with one of the triple-zeta SR basis sets. SettingNGRIDS 5 as above is useful for accelerating calculations with the MOLOPT basis sets.-T
Jiapeng Liu
Oct 1, 2019, 11:00:02 PM10/1/19
to cp2k
Hi Travis,
I just tried with your suggested script on my computer, but I got following errors
Program received signal SIGFPE: Floating-point exception - erroneous arithmetic operation.
Backtrace for this error:
Program received signal SIGFPE: Floating-point exception - erroneous arithmetic operation.
For your reference, I just installed cp2k on Ubuntu 18.04 by sudo apt-get install -y cp2k and cp2k version is 5.1 with cp2k.popt. Can you shed some light on this problem, thanks.
Regards,
Reply all
Reply to author
Forward
Message has been deleted
0 new messages