IC-QMMM with single charge in front of a metal plane: open boundary corrections

Katharina Doblhoff-Dier
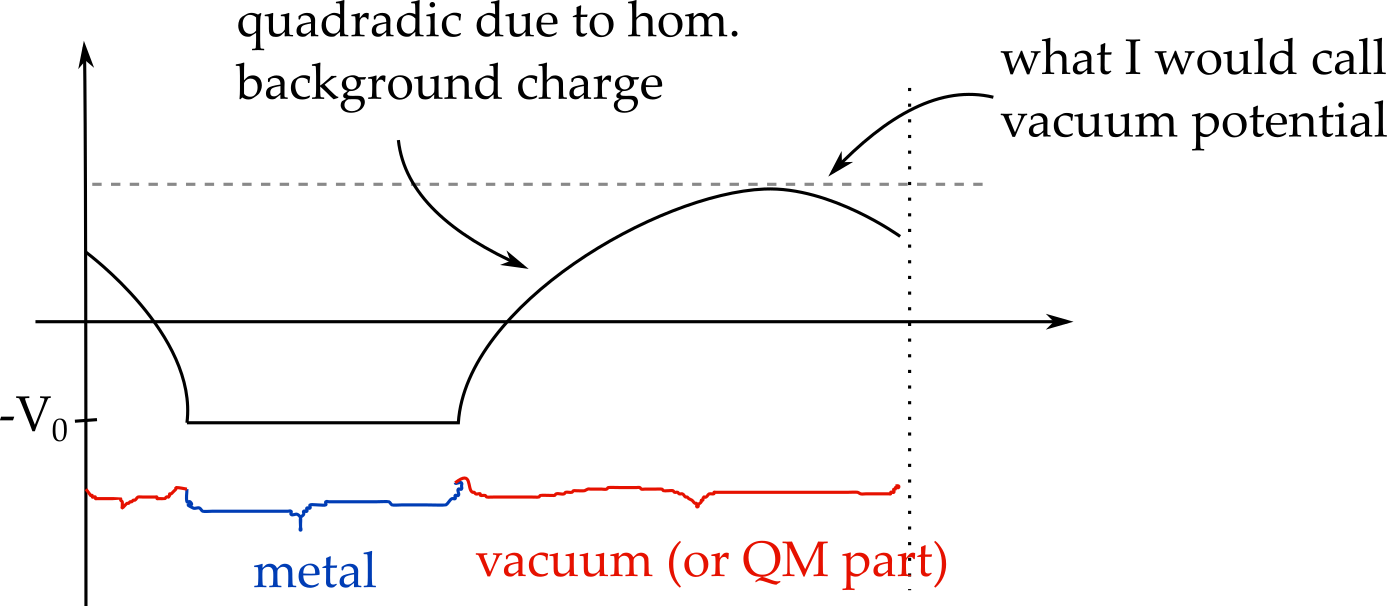
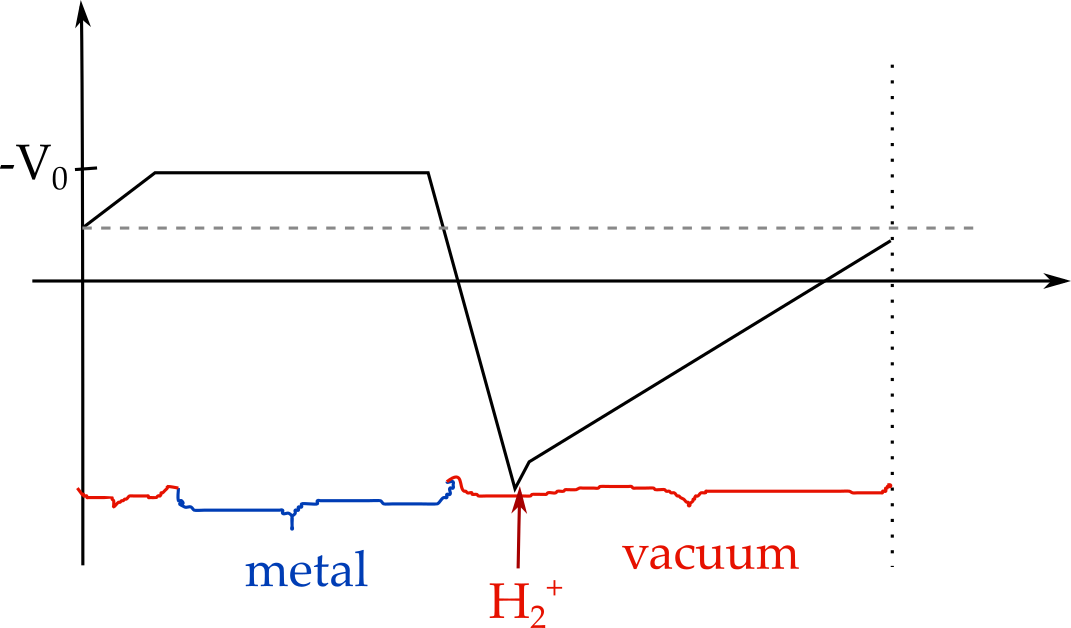
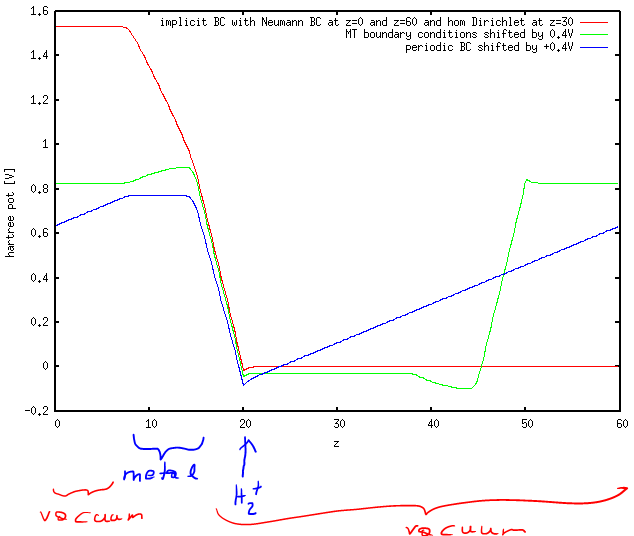
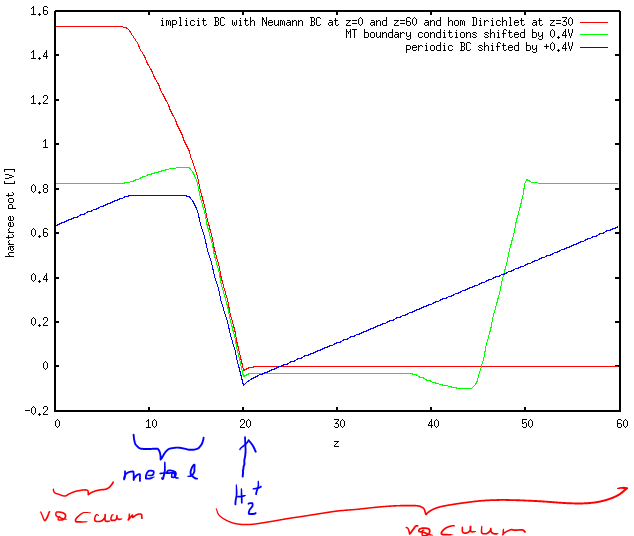
The corresponding V_0 (optimized such that Q=-1) were 1.11V for IMPLICIT boundary conditions, 1.24V for MT boundary conditions and -0.37V for periodic boundary conditions. While for periodic boundary conditions (blue line) this can be seen to correspond to the potential in the metal part, this is not the case for MT boundary conditions (green), where the potential in the metal varies from about -0.4 to -0.5 (remember that I shifted the curves by 0.4 volt) and for IMPLICIT boundary conditions. Overall, to me, it looks as if the IC-QMMM method was ignoring the boundary conditions and optimizing the charges for the periodic boundary conditions and then keeping them fixed no matter what I set as POISSON_SOLVER. In the MT boundary condition case (geen) we can thus see the influence of the charges shieding the (spurious) field in periodic boundary conditions (hence the slope in the metal part, which has the same slope as the average field in the periodic solver).
Finally, I decided that, in principle, it should be possible to find an energy minimum when Q_image=-Q_QM (as also shown in the original paper by Siepmann and Sprik). With none of the boundary conditions could I find this minimum correctly. However, here comes my non-understanding of Eq. 4 in the paper by Golze, Iannuzzi, ..., and Hutter (https://pubs-acs-org.ezproxy.leidenuniv.nl:2443/doi/10.1021/ct400698y) into play: Here, the energy is written as:
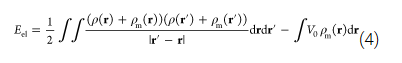
I would have thought this to be a grand canonical energy (grand canonical only in the charges on the metal) expression, where the last term accounts for the -N_i*mu_i term. Again, this does not seem physical to me if I think of a capacitor (or a charge+image charge in a box that is periodic in x and y, as I would then expect a correction for the charges in the QM region too (unless the vacuum potential on the QM side is zero and open boundary conditions are used, but likely this is wrong and this may be where my entire confusion starts.
So summarizing, this boils down to a few questions:
1.) can the IC-QMMM method be combined with poisson solvers other than periodic? If so, how? I simply set the poisson solver in the MM and the DFT part.
2.) What do I need to do in order to find an energy minimum for Q_image=-Q_QM?
3.) What is the meaning of V_0 and why is it substracted in the energy expression.

Dorothea Golze
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/5774878e-630c-4699-a67e-5040897c1b2do%40googlegroups.com.
Katharina Doblhoff-Dier
I am not sure if I understand your question correctly
and what your computational setup is.
&FORCE_EVAL
METHOD QMMM
&DFT
CHARGE 1
LSD
BASIS_SET_FILE_NAME BASIS_MOLOPT
POTENTIAL_FILE_NAME GTH_POTENTIALS
&POISSON
&EWALD
EWALD_TYPE ewald
ALPHA .44
GMAX 21
&END EWALD
POISSON_SOLVER MT
PERIODIC XY
&END POISSON
&MGRID
COMMENSURATE
CUTOFF 300
NGRIDS 5
&END MGRID
&QS
METHOD GPW
EXTRAPOLATION ASPC
EXTRAPOLATION_ORDER 3
&END QS
&SCF
MAX_SCF 300
SCF_GUESS ATOMIC
# SCF_GUESS RESTART
EPS_SCF 1.0E-6
&OT
PRECONDITIONER FULL_SINGLE_INVERSE
MINIMIZER DIIS
&END
&END SCF
&XC
&XC_FUNCTIONAL PBE
&END XC_FUNCTIONAL
&VDW_POTENTIAL
DISPERSION_FUNCTIONAL PAIR_POTENTIAL
&PAIR_POTENTIAL
TYPE DFTD3
CALCULATE_C9_TERM .TRUE.
REFERENCE_C9_TERM
PARAMETER_FILE_NAME dftd3.dat
REFERENCE_FUNCTIONAL PBE
R_CUTOFF [angstrom] 16.0
&END PAIR_POTENTIAL
&END VDW_POTENTIAL
&END XC
&PRINT
&V_HARTREE_CUBE
&END
&END
&END DFT
&MM
&FORCEFIELD
&CHARGE
ATOM Au
CHARGE 0
&END CHARGE
&CHARGE
ATOM H
CHARGE 0
&END CHARGE
&SPLINE
EPS_SPLINE 1.E-5
#EMAX_SPLINE 2.0
&END
&NONBONDED
&EAM
atoms Au Au
PARM_FILE_NAME Au.pot
&END EAM
&LENNARD-JONES
atoms Au H
EPSILON 0.0
SIGMA 3.166
RCUT 15
&END LENNARD-JONES
&LENNARD-JONES
atoms H H
EPSILON 0.0
SIGMA 3.166
RCUT 15
&END LENNARD-JONES
&END
&END FORCEFIELD
&POISSON
&EWALD
EWALD_TYPE ewald
ALPHA .44
GMAX 21
&END EWALD
POISSON_SOLVER MT
PERIODIC XY
&END POISSON
&END MM
&QMMM
CENTER NEVER
&CELL
ABC [angstrom] 34.6055 29.9693 60.0
PERIODIC XY
&END CELL
&QM_KIND H
MM_INDEX 577..578
&END QM_KIND
&IMAGE_CHARGE
EXT_POTENTIAL 1.24534
MM_ATOM_LIST 1..576
WIDTH 3.5
&END IMAGE_CHARGE
&PRINT
&IMAGE_CHARGE_INFO
&END
&END
&END QMMM
&SUBSYS
&CELL
ABC [angstrom] 34.6055 29.9693 60.0
PERIODIC XY
&END CELL
&TOPOLOGY
COORD_FILE_NAME Au_gua_image_dampFunc-opt.xyz
COORDINATE xyz
&END
&KIND H
BASIS_SET DZVP-MOLOPT-SR-GTH
POTENTIAL GTH-PBE-q1
&END KIND
&END SUBSYS
&END FORCE_EVAL
&GLOBAL
PROJECT Au_gua_image_dampFunc
RUN_TYPE energy
&END GLOBALKatharina Doblhoff-Dier
red: IMPLICIT poissn solver with Neumann BC at z=0 and z=60 and homogeneous Dirichlet BC at z=30green: MT (Martyna-Tuckerman) poisson solver (shifted by 0.4V)blue: normal Ewald summation (shifted by 0.4V)
Dorothea Golze
The constant potential condition is a hard constraint for the average. I tested with the standard periodic solver (for periodic systems) and the MT solver (for a cluster-type of approach).
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/168440d5-5b57-4075-900e-876a99f8e437o%40googlegroups.com.