SCF convergence issues
219 views
Skip to first unread message

Ryan Rogers
Oct 26, 2020, 5:07:46 PM10/26/20
to cp...@googlegroups.com
Dear CP2K community,
I am having issues in DFT QM/MM force calculations on molecular crystals of paracetamol (acetaminophen). I am describing here the two problems I most often experience.
I am currently unable to identify the cause or any pattern in the problems I encounter.
The root of the problems could be something other than what I identify below; I am pointing out the problematic features in the output that are most obviously to me. All input and output files are included.
1. Total energy falls into "hole" and never converges. (CP2K_problemTotalE_conf_0636.tar.gz)
Personal experience tells me to expect a "Total energy" for these systems on the order of -2,000 (Hartree) and a "Hartree energy" on the order of +2,000 (Hartree).
In these jobs, I find an initial "Hartree energy" on the order of >-10,000 (Hartree), which appears to send the SCF wavefunction optimization down a path of non-convergence, in which the "Total energy" can easily become on the order of -100,000 (Hartree) before I kill the job.
2. Total charge density on grids grows too large. (CP2K_problemEGrids_conf_0623.tar.gz)
In these jobs, the Total energy looks reasonable, and the Convergence looks promising in the few SCF cycles of steps.
However, the total Change never drops below my threshold, and eventually the "Total charge density on r-space/g-space grids" becomes much too large.
My configurations are extracted from MD trajectories, so the atoms have perturbations from their perfect crystal positions. One confusing observation is that very similar QM/MM configurations selected from other frames of the same trajectory often have no problems.
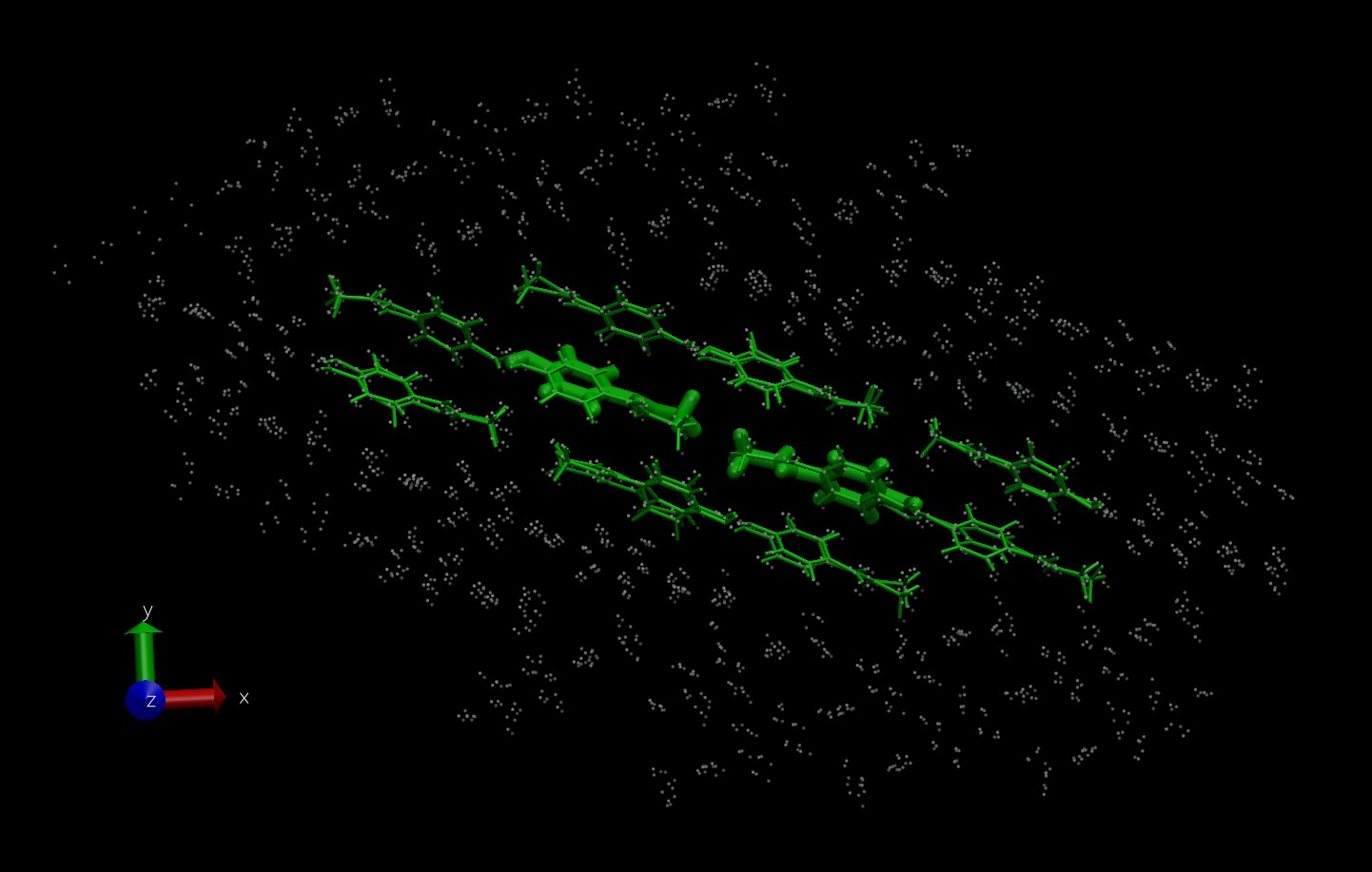
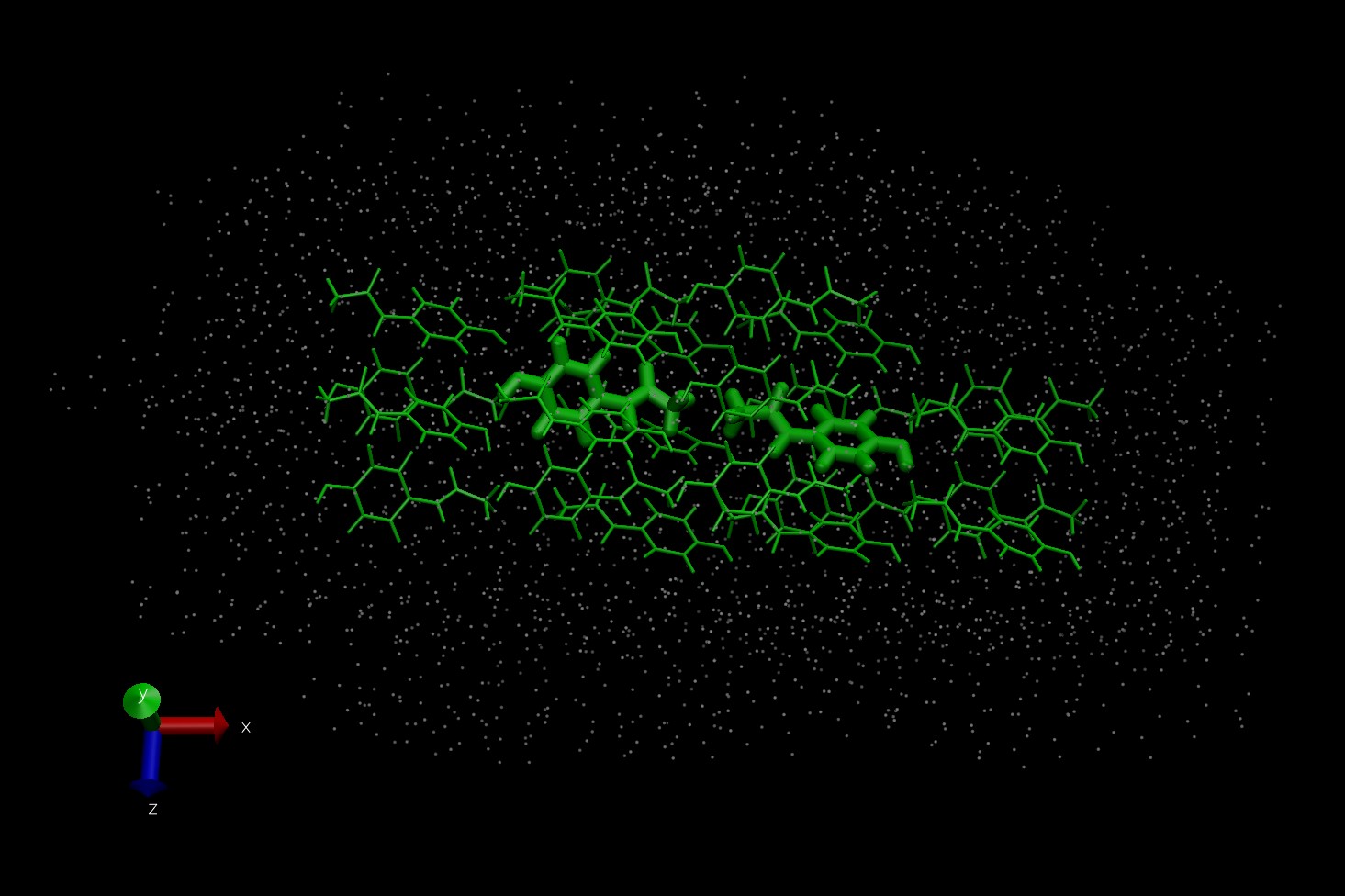
My configurations are constructed from a cluster of several molecules in the QM region with usually another layer usually 1-2 molecules thick making up the MM region. (In the attached sample images, the size of the stick molecules alludes to a larger/smaller basis set used, while the MM atoms are denoted as points.) Because I am not including integer numbers of unit cells, I am not using PBC.
Any advice about both/either problem will be greatly appreciated!
Ryan Rogers
Oct 29, 2020, 2:11:42 PM10/29/20
to cp2k
I might add that I have not been able to identify any obvious problems with the configurations (e.g. overlapping or too close atoms, etc.) when I encounter these errors.

Marcella Iannuzzi
Nov 2, 2020, 3:55:28 AM11/2/20
to cp2k
Dear Ryan Rogers
There is apparently a problem with the conservation of the charge on the QM grid.
Did you try to run a single point energy calculation for the QM part alone?
Kind regards
Marcella

{kind=link}
{kind=link}
{kind=link}
Женя Елизарова
Nov 5, 2020, 9:29:42 AM11/5/20
to cp2k
Dear Marcella Iannuzzi
I've just started to explore opportunities of the cp2k package. I've done some tutorials about single-point calculations of ethane molecule, QM/MM simulations, and some more. I am very interested in single point energy calculation for the QM part of the system. As I understood, I have to define the force_eval section (method - quickstep), and also I have to define subsections: dft, subsys, qmmm. Did I understand correctly? Also, i have some questions.
Do I have to define the MM subsection? In subsys section, I should define the whole system? How to define for which part of the system run a single point calculation?
Could you help me, please?
Best wishes,
Evgenia Elizarova
понедельник, 2 ноября 2020 г. в 11:55:28 UTC+3, Marcella Iannuzzi:
Marcella Iannuzzi
Nov 5, 2020, 11:08:44 AM11/5/20
to cp2k
Dear Evgenia Elizarova
Is this question related to the previous posts in this conversation?
If yes, what I meant is to remove all the MM part and just carry out a DFT calculation of the QM part.
Regards
Marcella
Женя Елизарова
Nov 5, 2020, 11:20:21 AM11/5/20
to cp2k
I see.
Actually, I would like to know is it possible to run a single point energy calculation of the QM subsystem without the removal of the MM part, not in relation to the previous posts (in general)?
Best wishes,
Evgenia
четверг, 5 ноября 2020 г. в 19:08:44 UTC+3, Marcella Iannuzzi:
Ryan Rogers
Nov 13, 2020, 6:25:51 PM11/13/20
to cp...@googlegroups.com
Dear Marcella,
Thank you very much for your suggestion. I tested removing the MM region, but unfortunately see similar behavior.
My configurations are generated from pure MM simulations with a custom force field which could possibly be allowing certain atoms to get a little too close. However, as the thermalized atom positions only vary (for the most part) by a few tenths of an Angstrom, no problematic close contacts can be found visually.
I've pasted below an example of the first SCF cycle from such a job, showing that the "Total energy" starts off much lower than I'm expecting (usually on the order of -2,000 +/- 500). During the SCF, the "Total energy" drops very low. When a job like this is allowed to continue, it won't crash on its own, but the "Total charge density on r-space grids" will become too large after 2-3 SCF cycles.
Is there any advice about how to handle atoms that are potentially "close" but not chemically "wrong"? These could potentially include hydrogen-bonding pairs like O-H, N-H, etc.
Any help is greatly appreciated in advance!
################################################################################
SCF WAVEFUNCTION OPTIMIZATION
----------------------------------- OT ---------------------------------------
Minimizer : DIIS : direct inversion
in the iterative subspace
using 7 DIIS vectors
safer DIIS on
Preconditioner : FULL_ALL : diagonalization, state selective
Precond_solver : DEFAULT
stepsize : 0.15000000 energy_gap : 0.00100000
eps_taylor : 0.10000E-15 max_taylor : 4
----------------------------------- OT ---------------------------------------
Step Update method Time Convergence Total energy Change
------------------------------------------------------------------------------
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
1 OT DIIS 0.15E+00 66.7 0.13322879 -8451.7456190410 -8.45E+03
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
2 OT DIIS 0.15E+00 65.4 0.13364185 -10643.9042780022 -2.19E+03
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
3 OT DIIS 0.15E+00 65.7 0.15497766 -13025.7969156521 -2.38E+03
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
4 OT DIIS 0.15E+00 65.5 0.14868108 -14464.8777105315 -1.44E+03
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
5 OT SD 0.15E+00 65.5 0.17759857 -14843.1753581151 -3.78E+02
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
6 OT DIIS 0.15E+00 65.8 0.32256964 -13383.1417204580 1.46E+03
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
7 OT DIIS 0.15E+00 65.6 0.19569799 -14813.0440620911 -1.43E+03
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
8 OT DIIS 0.15E+00 65.3 0.24452346 -15865.0025958057 -1.05E+03
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
9 OT DIIS 0.15E+00 65.5 0.26678603 -14795.2757334147 1.07E+03
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
10 OT DIIS 0.15E+00 65.7 0.36307397 -17004.3658543152 -2.21E+03
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
11 OT DIIS 0.15E+00 65.6 0.48782211 -17176.2139668702 -1.72E+02
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
12 OT DIIS 0.15E+00 65.5 0.45773260 -15669.9100288352 1.51E+03
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
13 OT DIIS 0.15E+00 65.5 0.66194382 -18842.9056350951 -3.17E+03
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
14 OT DIIS 0.15E+00 65.6 0.63269527 -16132.4918799441 2.71E+03
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
15 OT DIIS 0.15E+00 65.7 0.73064928 -16834.2508612376 -7.02E+02
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
16 OT DIIS 0.15E+00 65.5 1.15829587 -27460.2661838299 -1.06E+04
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
17 OT DIIS 0.15E+00 65.7 0.70782891 -15281.5935133825 1.22E+04
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
18 OT DIIS 0.15E+00 65.7 0.98480536 -20548.0516400897 -5.27E+03
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
19 OT DIIS 0.15E+00 65.6 1.39954759 -33842.8143338371 -1.33E+04
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
20 OT DIIS 0.15E+00 65.6 1.33809404 -29354.7460547364 4.49E+03
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
21 OT DIIS 0.15E+00 65.4 1.62600866 -36147.2741396100 -6.79E+03
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
22 OT DIIS 0.15E+00 65.6 1.21990609 -24956.2600892884 1.12E+04
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
23 OT DIIS 0.15E+00 65.6 1.32400959 -27440.1826004675 -2.48E+03
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
24 OT DIIS 0.15E+00 65.5 1.81006425 -42812.1680574805 -1.54E+04
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
25 OT DIIS 0.15E+00 65.7 0.89367434 -18701.9345340245 2.41E+04
Leaving inner SCF loop after reaching 25 steps.
Electronic density on regular grids: -1218.0000000005 -0.0000000005
Core density on regular grids: 1217.9999999992 -0.0000000008
Total charge density on r-space grids: -0.0000000013
Total charge density g-space grids: -0.0000000013
Overlap energy of the core charge distribution: 0.00009990728015
Self energy of the core charge distribution: -4772.82460287547656
Core Hamiltonian energy: 2367.11980010682919
Hartree energy: -15786.54494725671611
Exchange-correlation energy: -509.06361063631039
Dispersion energy: -0.62127327012014
QM/MM Electrostatic energy: 0.00000000000000
Total energy: -18701.93453402451269
outer SCF iter = 1 RMS gradient = 0.89E+00 energy = -18701.9345340245
################################################################################
SCF WAVEFUNCTION OPTIMIZATION
----------------------------------- OT ---------------------------------------
Minimizer : DIIS : direct inversion
in the iterative subspace
using 7 DIIS vectors
safer DIIS on
Preconditioner : FULL_ALL : diagonalization, state selective
Precond_solver : DEFAULT
stepsize : 0.15000000 energy_gap : 0.00100000
eps_taylor : 0.10000E-15 max_taylor : 4
----------------------------------- OT ---------------------------------------
Step Update method Time Convergence Total energy Change
------------------------------------------------------------------------------
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
1 OT DIIS 0.15E+00 66.7 0.13322879 -8451.7456190410 -8.45E+03
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
2 OT DIIS 0.15E+00 65.4 0.13364185 -10643.9042780022 -2.19E+03
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
3 OT DIIS 0.15E+00 65.7 0.15497766 -13025.7969156521 -2.38E+03
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
4 OT DIIS 0.15E+00 65.5 0.14868108 -14464.8777105315 -1.44E+03
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
5 OT SD 0.15E+00 65.5 0.17759857 -14843.1753581151 -3.78E+02
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
6 OT DIIS 0.15E+00 65.8 0.32256964 -13383.1417204580 1.46E+03
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
7 OT DIIS 0.15E+00 65.6 0.19569799 -14813.0440620911 -1.43E+03
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
8 OT DIIS 0.15E+00 65.3 0.24452346 -15865.0025958057 -1.05E+03
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
9 OT DIIS 0.15E+00 65.5 0.26678603 -14795.2757334147 1.07E+03
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
10 OT DIIS 0.15E+00 65.7 0.36307397 -17004.3658543152 -2.21E+03
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
11 OT DIIS 0.15E+00 65.6 0.48782211 -17176.2139668702 -1.72E+02
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
12 OT DIIS 0.15E+00 65.5 0.45773260 -15669.9100288352 1.51E+03
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
13 OT DIIS 0.15E+00 65.5 0.66194382 -18842.9056350951 -3.17E+03
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
14 OT DIIS 0.15E+00 65.6 0.63269527 -16132.4918799441 2.71E+03
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
15 OT DIIS 0.15E+00 65.7 0.73064928 -16834.2508612376 -7.02E+02
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
16 OT DIIS 0.15E+00 65.5 1.15829587 -27460.2661838299 -1.06E+04
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
17 OT DIIS 0.15E+00 65.7 0.70782891 -15281.5935133825 1.22E+04
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
18 OT DIIS 0.15E+00 65.7 0.98480536 -20548.0516400897 -5.27E+03
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
19 OT DIIS 0.15E+00 65.6 1.39954759 -33842.8143338371 -1.33E+04
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
20 OT DIIS 0.15E+00 65.6 1.33809404 -29354.7460547364 4.49E+03
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
21 OT DIIS 0.15E+00 65.4 1.62600866 -36147.2741396100 -6.79E+03
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
22 OT DIIS 0.15E+00 65.6 1.21990609 -24956.2600892884 1.12E+04
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
23 OT DIIS 0.15E+00 65.6 1.32400959 -27440.1826004675 -2.48E+03
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
24 OT DIIS 0.15E+00 65.5 1.81006425 -42812.1680574805 -1.54E+04
Adding QM/MM electrostatic potential to the Kohn-Sham potential.
25 OT DIIS 0.15E+00 65.7 0.89367434 -18701.9345340245 2.41E+04
Leaving inner SCF loop after reaching 25 steps.
Electronic density on regular grids: -1218.0000000005 -0.0000000005
Core density on regular grids: 1217.9999999992 -0.0000000008
Total charge density on r-space grids: -0.0000000013
Total charge density g-space grids: -0.0000000013
Overlap energy of the core charge distribution: 0.00009990728015
Self energy of the core charge distribution: -4772.82460287547656
Core Hamiltonian energy: 2367.11980010682919
Hartree energy: -15786.54494725671611
Exchange-correlation energy: -509.06361063631039
Dispersion energy: -0.62127327012014
QM/MM Electrostatic energy: 0.00000000000000
Total energy: -18701.93453402451269
outer SCF iter = 1 RMS gradient = 0.89E+00 energy = -18701.9345340245
################################################################################
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/0c182814-c0e4-42f2-84e0-cae9bb11377an%40googlegroups.com.
Marcella Iannuzzi
Nov 16, 2020, 6:52:45 AM11/16/20
to cp2k
The behaviour of the SCF is indeed very bad.
If possible, I would try to focus on the QM part only, simplify the model as much as you can, for instance using only one type of basis sets, and get it converged.
Kind regards
Marcella
Ryan Rogers
Nov 16, 2020, 4:23:53 PM11/16/20
to cp...@googlegroups.com
Dear Marcella,
I have done as you suggested, and tested a few different basis sets on a system with no MM charges, putting the same basis set on all QM atoms. In short, none of the basis sets I've tested so far seem to impact the SCF behavior.
I am using the MOLOPT basis sets with pseudopotentials in my QM region.
Under the hypothesis that there could be some unwanted charge transfer occurring in my system, I tested the smallest basis (MOLOPT SZV-SR), but to no avail.
Under the hypothesis that any possible charge transfer might need extra basis functions to be properly described/converged, I also tested the largest basis (MOLOPT TZV2PX); also to no avail.
Do you think there is any chance a molecular crystal (with strong hydrogen bond networks) could benefit from smearing?
I am admittedly not very familiar with different smearing methods, but know that they are mostly only prescribed for metals/conductor systems.
You received this message because you are subscribed to a topic in the Google Groups "cp2k" group.
To unsubscribe from this topic, visit https://groups.google.com/d/topic/cp2k/_W8iwo7KP40/unsubscribe.
To unsubscribe from this group and all its topics, send an email to cp2k+uns...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/cp2k/979b9ceb-46be-4c8f-b2da-41d912d2aa01n%40googlegroups.com.
Marcella Iannuzzi
Nov 17, 2020, 3:26:46 AM11/17/20
to cp2k
Dear Ryan
I don't know the electronic properties of your system. I would be surprised if it has a metallic character.
There might be some problem of charge delocalisation at the boundaries of the QM region.
Can you simulate with DFT only the isolated single molecule and the unit cell of the molecular crystal with PBC?
Best
Marcella
Reply all
Reply to author
Forward
0 new messages