Biodiverse for microbial ecology data
58 views
Skip to first unread message

Rebecca Prescott
Jan 5, 2021, 6:23:20 PM1/5/21
to Biodiverse Users
Aloha,
I have a dataset that are microbial 16S rRNA sequences from a number of caves in Hawaii. I am interested using randomization routines, NTI, etc...to look at community structure endemism levels within these sites. We have spatial coordinates for each cave, but typically this data is listed as samples or sites (caves). First question: do I have to have x and y coordinates in the base data file? If not, do you have an example file of something that does not have that dat included? I do have a rooted phylogenetic tree (.tre file) and I have the abundance (frequency) of each given sequence in a sample. There are multiple samples within a cave.
Just wondering if I am barking up a tree I can use! :) I think the maps would be great tools maybe for truly large scale microbial data, but not sure it will work for 10 caves.
Thanks in advance for any assistance!
Cheers
Becks

Shawn Laffan
Jan 5, 2021, 7:46:04 PM1/5/21
to biodiver...@googlegroups.com
Hello Becks,
Bidiverse will work for non-spatial data. The main caveat is that you cannot run spatial conditions that use distances.
An example is where we used word lists to analyse language distributions in Jones & Laffan (2006) - https://doi.org/10.1111/j.1467-968X.2008.00209.x
To analyse the data you can leave the spatial conditions as the default "sp_self_only()" (for spatial analyses) and "sp_select_all()" for cluster and region group analyses. There are also text matching conditions that could be used such as sp_match_regex() and sp_match_text() - https://github.com/shawnlaffan/biodiverse/wiki/SpatialConditions
The plots of the results will be based on a natural sort of the group names, with one cell per group. This can be difficult to interpret for large amounts of data, but for 10 items should not be an issue. (More details about natural sorts are at https://en.wikipedia.org/wiki/Natural_sort_order ).
To import the data you can specify "text_group" when allocating column types.
An example structure would look like this, with as many other columns as you need (i.e. you might have separate columns for family, genus and species) or perhaps some that are not needed. You can also have more than one text column to define the groups.
cave_name,spp_name
cave1,sp1
cave1,sp2
cave2,sp1
cave2,sp3
In terms of endemism levels, be aware that the ranges will be in units of caves. If they are similar sizes then it is not a big issue. Ten caves is also unlikely to span the full range of the taxa being studied, but such undersampling affects almost all endemism analyses.
WRT the randomisations and NRI/NTI, the NRI and NTI indices include a randomisation process so do not need to then be further randomised. If you want to use some of the constrained randomisations with text based conditions then use the MPD/MNTD indices directly.
Regards,
Shawn.
Bidiverse will work for non-spatial data. The main caveat is that you cannot run spatial conditions that use distances.
An example is where we used word lists to analyse language distributions in Jones & Laffan (2006) - https://doi.org/10.1111/j.1467-968X.2008.00209.x
To analyse the data you can leave the spatial conditions as the default "sp_self_only()" (for spatial analyses) and "sp_select_all()" for cluster and region group analyses. There are also text matching conditions that could be used such as sp_match_regex() and sp_match_text() - https://github.com/shawnlaffan/biodiverse/wiki/SpatialConditions
The plots of the results will be based on a natural sort of the group names, with one cell per group. This can be difficult to interpret for large amounts of data, but for 10 items should not be an issue. (More details about natural sorts are at https://en.wikipedia.org/wiki/Natural_sort_order ).
To import the data you can specify "text_group" when allocating column types.
An example structure would look like this, with as many other columns as you need (i.e. you might have separate columns for family, genus and species) or perhaps some that are not needed. You can also have more than one text column to define the groups.
cave_name,spp_name
cave1,sp1
cave1,sp2
cave2,sp1
cave2,sp3
In terms of endemism levels, be aware that the ranges will be in units of caves. If they are similar sizes then it is not a big issue. Ten caves is also unlikely to span the full range of the taxa being studied, but such undersampling affects almost all endemism analyses.
WRT the randomisations and NRI/NTI, the NRI and NTI indices include a randomisation process so do not need to then be further randomised. If you want to use some of the constrained randomisations with text based conditions then use the MPD/MNTD indices directly.
Regards,
Shawn.
--
You received this message because you are subscribed to the Google Groups "Biodiverse Users" group.
To unsubscribe from this group and stop receiving emails from it, send an email to biodiverse-use...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/biodiverse-users/62a3eeaa-c1e3-4c53-a600-bb813b966f94n%40googlegroups.com.
Rebecca Prescott
Jan 5, 2021, 10:09:05 PM1/5/21
to Biodiverse Users
Aloha Shawn,
Thanks for the quick reply, and very helpful. I have attached a .tsv file that has the data in the format it is now (this is a subset so the file is not too big). You can see that its an abundance table. Columns are samples. We have 51 samples from across 10 caves, and . The rows are the sequence IDs - these are amplicon sequence variants (something similar to OTUs if those are familiar to you and ASVs are not). These IDs of numbers and letters are representative of a specific DNA sequence, and they match the tips of the phylogenetic tree that we have constructed for these same data. I did not attach it - its too big I think. We do have taxonomic identification for each sequence, but those taxon names will not match the phylogenetic tree, and they vary in terms of taxonomic level that a specific sequence is identified to (some are to genus, others are only to class). The tree branch lengths represent nucleotide changes.
The main question of interest is is there a higher or lower level of endemism than expected by chance in individual samples and/or each cave?
I would think we would need to keep the tip branch IDs and the base data file IDs the same? And not sure if we can or should keep the abundance data (i.e. the counts of each sequence in each sample)?
I am not sure if anyone has used Biodiverse with microbial sequence data - have not found a paper yet with that. Just thought it might be helpful for you to see how the data is structured now, to help sort how how to change it for Biodiverse appropriately. :) I think if I can get that correct, then I can work out the rest with some further reading of the guide, papers, etc..
Greatly appreciate you help and time!
Cheers
Becks
Shawn Laffan
Jan 6, 2021, 5:33:29 PM1/6/21
to biodiver...@googlegroups.com
Hello Becks,
Those data can be imported using matrix format. You will need to delete the current first line so the header comes first.
It would also be helpful to change the file name extension to txt so the file selection system shows it by default. Otherwise you need to change the "txt and csv files" widget at the bottom right to "all files". This is not essential, but does save some button clicking.
On the screen after the file selection, set the "data are in matrix form?" checkbox to on.
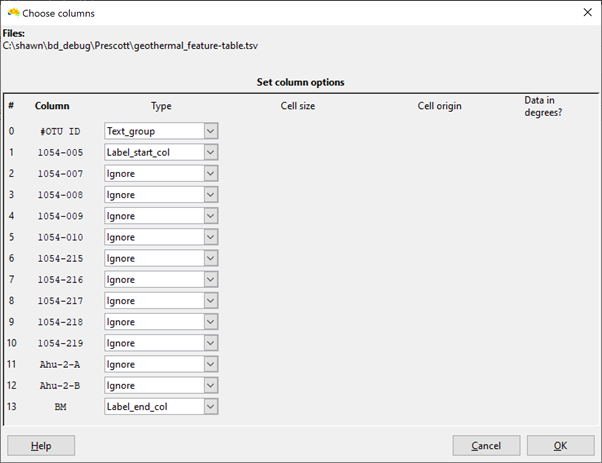
As your data are in a sites-as-columns format, they need to be imported in transposed form (OTUs as groups, caves as labels). When selecting the columns, set the OTU as text_group and the site columns as labels (see first screen shot below).
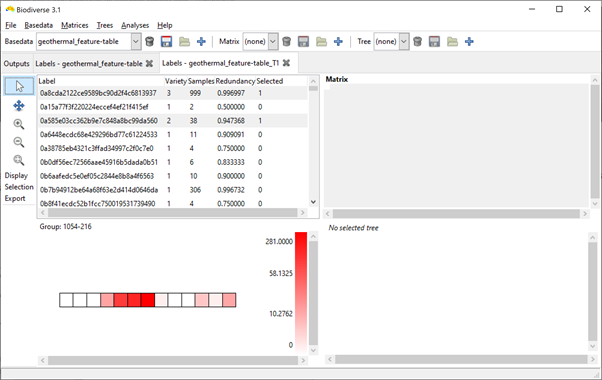
Once the data are imported, choose the Basedata > Transpose menu option to create a basedata with the caves as the groups and the OTUs as the labels (second screenshot shows what this will look like).
In regards to linking the basedata and the tree, Biodiverse knows nothing about the Linnean system of taxonomy - all it sees is text and numbers. This is why we refer to labels. (A similar principle applies to groups). So long as the labels in the basedata match the terminal names on the tree then the analyses will work. There is a remap system for cases where they do not match: https://biodiverse-analysis-software.blogspot.com/2017/04/matching-spatial-tree-matrix-and.html
WRT the abundance data, keep them. They have no effect on endemism analyses, and might become useful later if you want to run some of the abundance weighted indices.
Once you have it all imported then you can run the various indices, followed by randomisations as needed. Some of the RPD/RPE indices might also be useful. These use the topology of the tree which can be useful for a variety of reasons (see for example Mishler et al. 2014, 2020).
https://doi.org/10.1038/ncomms5473
https://doi.org/10.1111/jse.12590
I am not aware of direct use of microbial data with Biodiverse, but as I noted above it is all numbers. More generally, Phylogenetic Diversity (PD) has been used for such data in the past (as a start look for UniFrac, and also https://doi.org/10.1371/journal.pcbi.1002832 ). You can run whatever analyses you like with whatever data you like - the important differences are usually in how the data are interpreted (the first step checking they make sense, of course).
For further reading about Biodiverse, have a look at the blog and also the publications list.
https://biodiverse-analysis-software.blogspot.com/ (for posts on the randomisations, see https://biodiverse-analysis-software.blogspot.com/search/label/randomisations ).
https://github.com/shawnlaffan/biodiverse/wiki/PublicationsList
Please do ask if you have any further questions. Success stories are also always welcome.
Regards,
Shawn.
Those data can be imported using matrix format. You will need to delete the current first line so the header comes first.
It would also be helpful to change the file name extension to txt so the file selection system shows it by default. Otherwise you need to change the "txt and csv files" widget at the bottom right to "all files". This is not essential, but does save some button clicking.
On the screen after the file selection, set the "data are in matrix form?" checkbox to on.
As your data are in a sites-as-columns format, they need to be imported in transposed form (OTUs as groups, caves as labels). When selecting the columns, set the OTU as text_group and the site columns as labels (see first screen shot below).
Once the data are imported, choose the Basedata > Transpose menu option to create a basedata with the caves as the groups and the OTUs as the labels (second screenshot shows what this will look like).
In regards to linking the basedata and the tree, Biodiverse knows nothing about the Linnean system of taxonomy - all it sees is text and numbers. This is why we refer to labels. (A similar principle applies to groups). So long as the labels in the basedata match the terminal names on the tree then the analyses will work. There is a remap system for cases where they do not match: https://biodiverse-analysis-software.blogspot.com/2017/04/matching-spatial-tree-matrix-and.html
WRT the abundance data, keep them. They have no effect on endemism analyses, and might become useful later if you want to run some of the abundance weighted indices.
Once you have it all imported then you can run the various indices, followed by randomisations as needed. Some of the RPD/RPE indices might also be useful. These use the topology of the tree which can be useful for a variety of reasons (see for example Mishler et al. 2014, 2020).
https://doi.org/10.1038/ncomms5473
https://doi.org/10.1111/jse.12590
I am not aware of direct use of microbial data with Biodiverse, but as I noted above it is all numbers. More generally, Phylogenetic Diversity (PD) has been used for such data in the past (as a start look for UniFrac, and also https://doi.org/10.1371/journal.pcbi.1002832 ). You can run whatever analyses you like with whatever data you like - the important differences are usually in how the data are interpreted (the first step checking they make sense, of course).
For further reading about Biodiverse, have a look at the blog and also the publications list.
https://biodiverse-analysis-software.blogspot.com/ (for posts on the randomisations, see https://biodiverse-analysis-software.blogspot.com/search/label/randomisations ).
https://github.com/shawnlaffan/biodiverse/wiki/PublicationsList
Please do ask if you have any further questions. Success stories are also always welcome.
Regards,
Shawn.
To view this discussion on the web visit https://groups.google.com/d/msgid/biodiverse-users/59a62324-fc24-41d0-b3d3-fb49c45b1a1fn%40googlegroups.com.
Rebecca Prescott
Jan 11, 2021, 4:54:18 PM1/11/21
to biodiver...@googlegroups.com
Aloha Shawn,
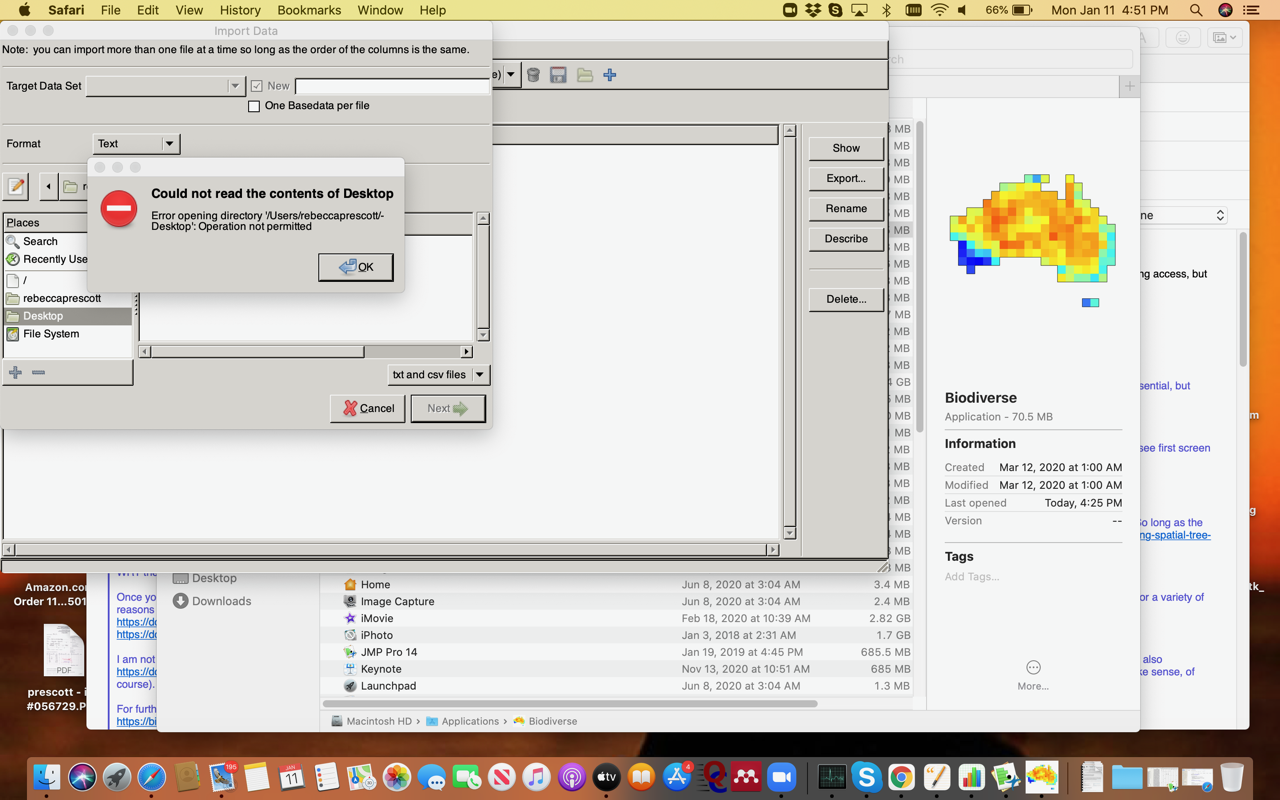
Thanks very much for the details below. I have run into a problem - I am using a Mac (OS Catalina 10.15.5). So far, I have not been able to get Biodiverse to be able to access the files. I did try going into security and giving access via “full access", but did not work. See the screen shot below. Do you have any recommendations to get Biodiverse access? If I have missed this somewhere in the documentation, my apologies!
Cheers
Becks
On Jan 6, 2021, at 5:32 PM, Shawn Laffan <shawn....@unsw.edu.au> wrote:
Hello Becks,
Those data can be imported using matrix format. You will need to delete the current first line so the header comes first.
It would also be helpful to change the file name extension to txt so the file selection system shows it by default. Otherwise you need to change the "txt and csv files" widget at the bottom right to "all files". This is not essential, but does save some button clicking.
On the screen after the file selection, set the "data are in matrix form?" checkbox to on.
As your data are in a sites-as-columns format, they need to be imported in transposed form (OTUs as groups, caves as labels). When selecting the columns, set the OTU as text_group and the site columns as labels (see first screen shot below).
Once the data are imported, choose the Basedata > Transpose menu option to create a basedata with the caves as the groups and the OTUs as the labels (second screenshot shows what this will look like).
In regards to linking the basedata and the tree, Biodiverse knows nothing about the Linnean system of taxonomy - all it sees is text and numbers. This is why we refer to labels. (A similar principle applies to groups). So long as the labels in the basedata match the terminal names on the tree then the analyses will work. There is a remap system for cases where they do not match: https://biodiverse-analysis-software.blogspot.com/2017/04/matching-spatial-tree-matrix-and.html
WRT the abundance data, keep them. They have no effect on endemism analyses, and might become useful later if you want to run some of the abundance weighted indices.
Once you have it all imported then you can run the various indices, followed by randomisations as needed. Some of the RPD/RPE indices might also be useful. These use the topology of the tree which can be useful for a variety of reasons (see for example Mishler et al. 2014, 2020).
https://doi.org/10.1038/ncomms5473
https://doi.org/10.1111/jse.12590
I am not aware of direct use of microbial data with Biodiverse, but as I noted above it is all numbers. More generally, Phylogenetic Diversity (PD) has been used for such data in the past (as a start look for UniFrac, and also https://doi.org/10.1371/journal.pcbi.1002832 ). You can run whatever analyses you like with whatever data you like - the important differences are usually in how the data are interpreted (the first step checking they make sense, of course).
For further reading about Biodiverse, have a look at the blog and also the publications list.
https://biodiverse-analysis-software.blogspot.com/ (for posts on the randomisations, see https://biodiverse-analysis-software.blogspot.com/search/label/randomisations ).
https://github.com/shawnlaffan/biodiverse/wiki/PublicationsList
Please do ask if you have any further questions. Success stories are also always welcome.
Regards,
Shawn.
<kfekgnpmcmbijfoc.png>
<fhkacplodcnlfapp.png>
--
You received this message because you are subscribed to a topic in the Google Groups "Biodiverse Users" group.
To unsubscribe from this topic, visit https://groups.google.com/d/topic/biodiverse-users/bC8AhXz7Gq4/unsubscribe.
To unsubscribe from this group and all its topics, send an email to biodiverse-use...@googlegroups.com.
To view this discussion on the web visit https://groups.google.com/d/msgid/biodiverse-users/908bdcab-90db-0e71-fed3-79c2162a2d6b%40unsw.edu.au.
Shawn Laffan
Jan 11, 2021, 6:42:43 PM1/11/21
to biodiver...@googlegroups.com
Hello Becks,
This looks like issue #778 - https://github.com/shawnlaffan/biodiverse/issues/778
Did you recently upgrade to Catalina? And does it only affect Biodiverse?
Regards,
Shawn.
This looks like issue #778 - https://github.com/shawnlaffan/biodiverse/issues/778
Did you recently upgrade to Catalina? And does it only affect Biodiverse?
Regards,
Shawn.
On 12/01/2021 8:54, Rebecca Prescott
wrote:
Aloha Shawn,
Thanks very much for the details below. I have run into a problem - I am using a Mac (OS Catalina 10.15.5). So far, I have not been able to get Biodiverse to be able to access the files. I did try going into security and giving access via “full access", but did not work. See the screen shot below. Do you have any recommendations to get Biodiverse access? If I have missed this somewhere in the documentation, my apologies!
CheersBecks
To view this discussion on the web visit https://groups.google.com/d/msgid/biodiverse-users/F902BC66-B85D-4704-A462-E90D3FB7FC4D%40gmail.com.
Rebecca Prescott
Jan 11, 2021, 6:52:33 PM1/11/21
to biodiver...@googlegroups.com
Aloha,
Thanks for link below!! Sorry I missed it. Will take a look and let ya know…:)
Cheers
Becks
To view this discussion on the web visit https://groups.google.com/d/msgid/biodiverse-users/47f08621-3ed6-100a-c734-78b3be16f413%40unsw.edu.au.
Shawn Laffan
Jan 11, 2021, 7:17:37 PM1/11/21
to biodiver...@googlegroups.com
The Biodiverse releases use a system that packages up all the
component files into a self-extracting zip file. On Macs this is
further packaged into the dmg file for installation.
The first run of any installation will be slow as it needs to unpack all of its files into a directory so they can then be accessed. This is cached so subsequent runs do not need to unpack the files. By default these files are unpacked into the temp directory.
The slow startup is likely because the system is cleaning up the temp directory, and so all the files need to be unpacked again each run. (This can also affect Windows users when tools like CCleaner are in use).
As for the issues opening files, I wonder if there are security issues with calling executable files from the temp directory.
If you are familiar with the command line then could you try running this line from a terminal? (Hopefully it works - I do not have a mac to work from right now).
PAR_GLOBAL_TEMP=${HOME}/bd_exe_files Biodiverse.app
That line will change the location the Biodiverse files are unpacked to so they are under your home directory instead of the temp directory. It will only change it for that invocation of the command - if it works then we can change some other settings to make it more permanent.
If it is the wrong call for Biodiverse then the file to call will be somewhere in the /Applications directory.
Regards,
Shawn.
The first run of any installation will be slow as it needs to unpack all of its files into a directory so they can then be accessed. This is cached so subsequent runs do not need to unpack the files. By default these files are unpacked into the temp directory.
The slow startup is likely because the system is cleaning up the temp directory, and so all the files need to be unpacked again each run. (This can also affect Windows users when tools like CCleaner are in use).
As for the issues opening files, I wonder if there are security issues with calling executable files from the temp directory.
If you are familiar with the command line then could you try running this line from a terminal? (Hopefully it works - I do not have a mac to work from right now).
PAR_GLOBAL_TEMP=${HOME}/bd_exe_files Biodiverse.app
That line will change the location the Biodiverse files are unpacked to so they are under your home directory instead of the temp directory. It will only change it for that invocation of the command - if it works then we can change some other settings to make it more permanent.
If it is the wrong call for Biodiverse then the file to call will be somewhere in the /Applications directory.
Regards,
Shawn.
To view this discussion on the web visit https://groups.google.com/d/msgid/biodiverse-users/D1E22595-2B4C-45E2-84E8-B9AB233673F8%40gmail.com.
-- Prof Shawn Laffan, FMSSANZ School of Biological, Earth and Environmental Sciences UNSW, Sydney 2052, Australia Tel +61 2 9385 8093 https://www.bees.unsw.edu.au/our-people/shawn-laffan https://shawnlaffan.github.io/biodiverse (free diversity analysis software) International Journal of Geographical Information Science http://www.tandf.co.uk/journals/ijgis UNSW CRICOS Provider Code 00098G
Rebecca Prescott
Jan 11, 2021, 8:04:18 PM1/11/21
to biodiver...@googlegroups.com
Aloha,
I installed several days ago, and moved to applications. So not sure this was related to unpacking the files or not? It does seem to have stopped once I changed the security settings just like in issue #778. However, that change did not allow access to the files on the desktop, or higher up directories.
I can try running the command below, but thought I would try downloading PC version and running in Parallels first?? As a work around…
Would love to sort out! :) Or I will have to find someone with a PC or different computer...
Cheers
Becks
To view this discussion on the web visit https://groups.google.com/d/msgid/biodiverse-users/8b8803a9-eeb1-fdeb-fa83-4d47ef66bfe4%40unsw.edu.au.
Rebecca Prescott
Jan 11, 2021, 8:31:08 PM1/11/21
to biodiver...@googlegroups.com
Aloha,
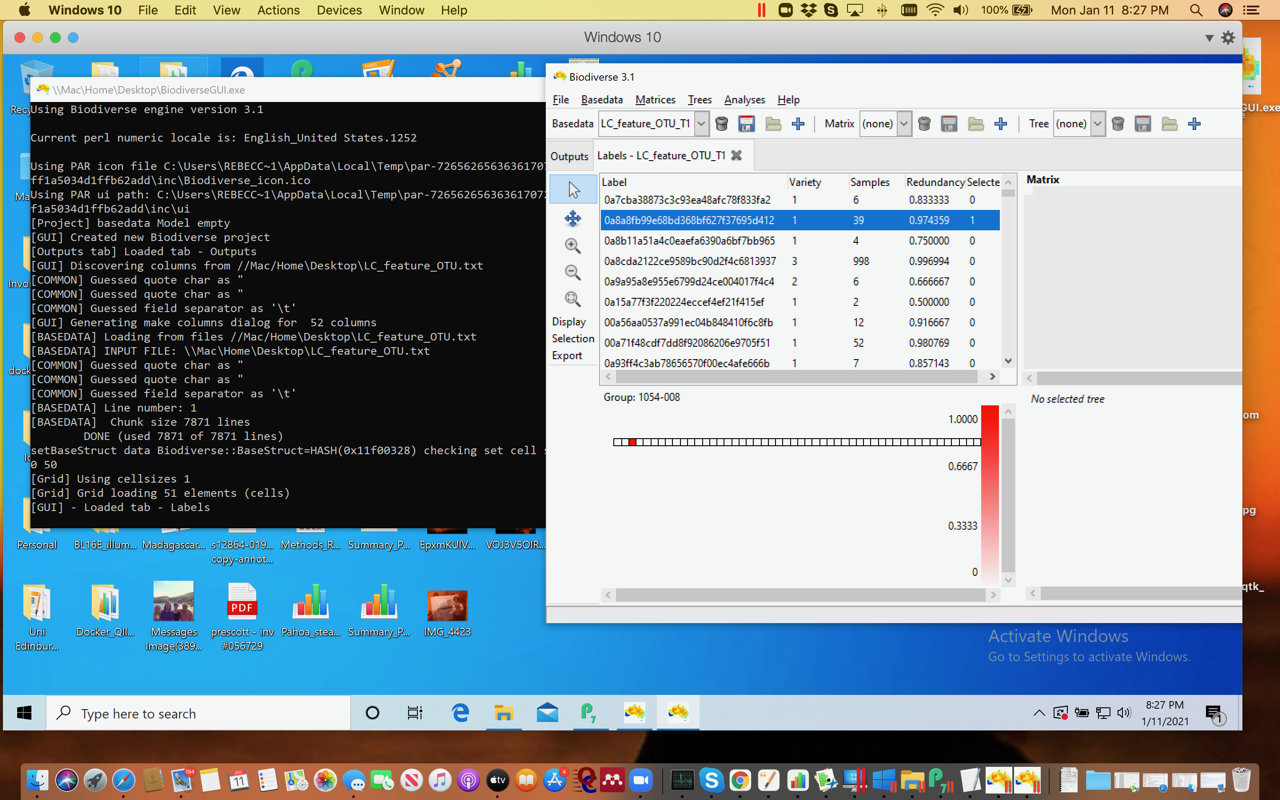
Just an update: using Parallels and downloading the .exe file from Github seems to have worked - not getting security warnings now. Although I may have made a mistake with the import of the file. But it can see the files and I could import…something. :) Stumbling my way through and need to re-read the directions a bit. These data have 7082 runs ( = to OTUs, or something like that!) and 51 columns. Any limits on size of file Biodiverse can handle?
To view this discussion on the web visit https://groups.google.com/d/msgid/biodiverse-users/8b8803a9-eeb1-fdeb-fa83-4d47ef66bfe4%40unsw.edu.au.
Shawn Laffan
Jan 11, 2021, 8:51:11 PM1/11/21
to biodiver...@googlegroups.com
Moving to applications is the process.
To clarify, are you able to open files in directories other than the Desktop?
If you are able to run the command then that would be very helpful.
There is also a log file that lives under the $HOME/Library directory (I think). You might need to use the finder to get it - look for a file called Biodiverse.log. If you can send that to me directly (rather than via the list) it would likely be helpful.
Regards,
Shawn.
To clarify, are you able to open files in directories other than the Desktop?
If you are able to run the command then that would be very helpful.
There is also a log file that lives under the $HOME/Library directory (I think). You might need to use the finder to get it - look for a file called Biodiverse.log. If you can send that to me directly (rather than via the list) it would likely be helpful.
Regards,
Shawn.
To view this discussion on the web visit https://groups.google.com/d/msgid/biodiverse-users/61FC75A1-FEDB-4778-A3A1-D9D3BC219A2D%40gmail.com.
Shawn Laffan
Jan 11, 2021, 8:54:01 PM1/11/21
to biodiver...@googlegroups.com
Good to hear you got it working via Parallels. I would still like
to get it working on Catalina, but those details be discussed on the
other sub-thread.
As for data set size, it is entirely a function of the machine you are running it on. For the North American flora work we had about 20,000 labels and 50,000 groups (Mishler et al. 2020, https://doi.org/10.1111/jse.12590 ).
Regards,
Shawn.
As for data set size, it is entirely a function of the machine you are running it on. For the North American flora work we had about 20,000 labels and 50,000 groups (Mishler et al. 2020, https://doi.org/10.1111/jse.12590 ).
Regards,
Shawn.
To view this discussion on the web visit https://groups.google.com/d/msgid/biodiverse-users/119D195D-FD3E-40BC-9EA9-5039E9DC3A01%40gmail.com.
Shawn Laffan
Jan 12, 2021, 8:25:10 PM1/12/21
to biodiver...@googlegroups.com
Just an update for the list following a debug session with Becks:
Files can be opened from directories other than Desktop, Documents etc. Storing Biodiverse files in a location such as
I'd appreciate it if there are any MacOS Catalina users who could try the above workarounds and report if they work or not.
Shawn.
Files can be opened from directories other than Desktop, Documents etc. Storing Biodiverse files in a location such as
/home/username/biodiverse
is one workaround.
The error does not occur when calling the script directly from a command terminal instead of the Finder:
/Applications/Biodiverse.app/Contents/MacOS/Biodiverse
I'd appreciate it if there are any MacOS Catalina users who could try the above workarounds and report if they work or not.
Shawn.
To view this discussion on the web visit https://groups.google.com/d/msgid/biodiverse-users/9595a89b-f3c0-220f-6e81-ddd325959b30%40unsw.edu.au.
Reply all
Reply to author
Forward
0 new messages