Quality filter non-multiplexed samples stitched with Pandaseq: output file empty!

Niccolo`
I am trying to analyse my 16S data coming from a run of Miseq PE 2x250. I submitted 6 samples and I have received from the sequencing center 6 fastq files (12 if we consider R1 and R2), so already demultiplexed and without any barcode file.
I have used Pandaseq to stitch R1 and R2 reads together and now I would like to run split_libraries_fastq.py to do quality filtering. This is the command I used:
split_libraries_fastq.py -i stitched.fastq --sample_id Y -o split_libraries/ -m Ymapfileforsplitlibr.txt -q20 --barcode_type 'not-barcoded'
The script works but the output fasta file is empty and in the log file it is written:
Quality filter results
Total number of input sequences: 448526
Barcode not in mapping file: 0
Read too short after quality truncation: 448526
Count of N characters exceeds limit: 0
Illumina quality digit = 0: 0
Barcode errors exceed max: 0
Result summary (after quality filtering)
Median sequence length: nan
MK3 0
Total number seqs written 0
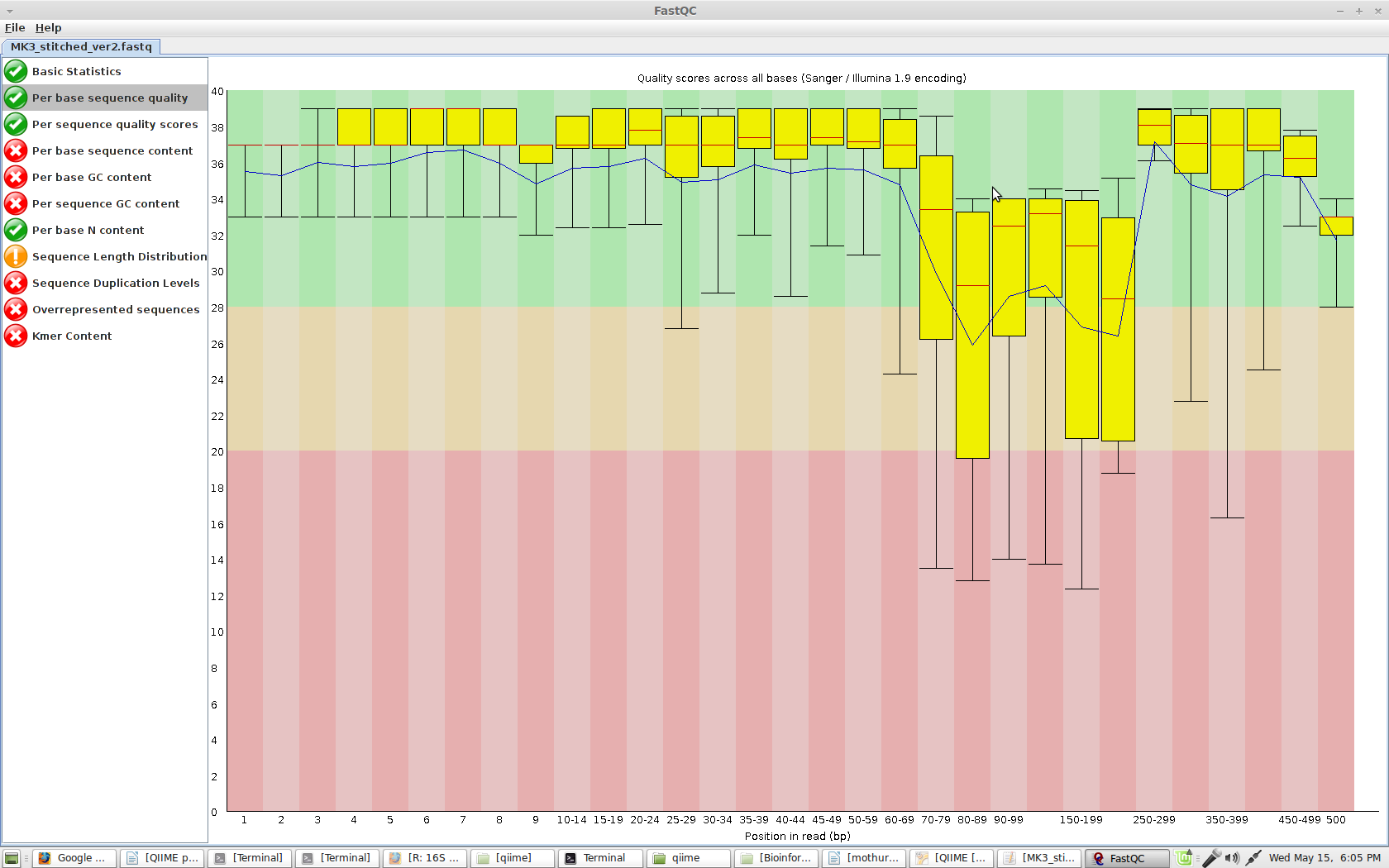
I am enclosing the FastQC screenshot of the stitched file: the qualiy drops a bit in the middle,but could that explain the problem? I tried to run split_libraries_fastq.py without -q20 but still I can keep only few reads.
Can anyone please help me to understand what is the problem?
I also read the discussion https://groups.google.com/forum/#!msg/qiime-forum/CO9EmR4FH58/vNuaaOyAv-cJ but I think my case is different since I don`t have the problem to combine the barcode file and the stitched file.
Just in case here is how the original file and the stitched files look like:
original
@M01168:27:000000000-A2Y50:1:1101:14172:1431 1:N:0:16
CTGGACCGTGTCTCAGTTCCAGTGTGGCTGGTCATCCTCTCAGACCAGCTAGAGATCGTCGGCTTGGTAGGCCTTTACCCCACCAACTACCTAATCCCACTTGGGCTCATCTTATGGCATATGGTCTTGCGATCCCATACTTTAATCTTTCGATATTACGCGGTATTAGCTACAGTTTCCCGTAGTTATCCCCCTCCATAAGCCAGATTCCCAAGCATTACTCACCCGTCCGC
+
GGGGHHHGGGFHHHGHHGHHHGHHHFFHHGGHHHHHHHHHHHHHHHHHHHHHGHHHHGHHGGGGGGGGHHGHHHHHGHHHHGGGHHGHHHHHHHHHHHHHHHHHHHHHHHHHHHHHEHHHHHHHHHHHGHGGHGGHHHGHHHHHHHGHHHHGGHGGHHHAAFGFGGHHHHHHHHGHHHHHHGHGGFFGGGFFFGGGGGGGGFFBFFGGGGGFFGEGFFFFFFFFFFFDD?AB;
@M01168:27:000000000-A2Y50:1:1101:16756:1431 1:N:0:16
GATGAACGCTAGCGGCAGGCCTAAAACATGCAAGTCGAGGGGCAAGGGGCTTCGGCCTCCTAGACCGGCGTACGGGTGCGTAACGCGTGTACAATCTACCTTTTACTACTGGATATCCCGGAGAAATTTGGATTAATACGGTATAGTATTATTTATAGGCATCTATTAATAATTAAAGATTTATTGGTAAAAGATGAGTACGCGTTCTATTAGTTAGTTGGAGTGGTAACG
+
GFGHHHHHGGGGHCCGCEG?FBAG3BFGFCFHHFBFEEEGGGGEGFEFG?/1FEF?EEGEHHHD?F@EEG?@/<AC/BAD/AD<?AC>D<0FGF1>1<F1<F<GGHDDF00C/DDDGHHFGGGGHH;FHHGGBFFFBB;EG:E.FF;CFBFFGGGBFFCBCCF0;0CFGGGGFFFFFFFFBFFBFFBBFFBFBEFFB;/B9.DB--9EFFFFF/:;99;FFBB9AA9BF/.
stitched
@M01168:27:000000000-A2Y50:1:1101:14172:1431:16
ATTGAACGCTGGCGGCAGGCTTAACACATGCAAGTCGAACGGTAGCAGGAAGTACTTGTTTTCTTGCTGACGAGTGGCGGACGGGTGAGTAATGCTTGGGAATCTGGCTTATGGAGGGGGATAACTACGGGAAACTGTAGCTAATACCGCGTAATATCGAAAGATTAAAGTATGGGATCGCAAGACCATATGCCATAAGATGAGCCAAAGTGGGATTAGGTAGTTGGTGGGGTAAAGGCCTACCAAGCCGACGATCTCTAGCTGGTCTGAGAGGATGACCAGCCACACTGGAACTGAGACACGGTCCAG
+
FFFHHHHHFFF?FFCCFFFFHHFH5FFFHFHHFFFHFEAFECEEEEFEC?FF@FFHFHFHFHHHHHHFFBFFFFFH:@@>>.;AA;=.B!>B0B0CAA.ACDD0B@CBBD0CBCCC>?A>BBBCACDD>-,:?C@CDDCCCDDCCCC@?@@,9A>:8:8@CCCCCCCCCC@CCCCCC@9@B>-8A-8AAACC.:CC88ACC.88AA!A-.!-8-!!:8.9.:8.:.:8C@CHFHHHHHFHHFFFFFFFFHHFHHHHFHHHHHHHHHHHHHHHHHHHHHFFHHFFHHHFHHHFHHFHHHFFFFHHHFFFF
@M01168:27:000000000-A2Y50:1:1101:16756:1431:16
CTGGTCCGTGTCTCAGTACCAGTGGGGGGGACATCCCTCTCAGAACCCCTATCTATCATCGCCTTGGTGTGCCGTTACCACTCCAACTAACTAATAGAACGCGTACTCATCTTTTACCAATAAATCTTTAATTATTAATAGATGCCTATAAATAATACTATACCGTATTAATCCAAATTTCTCCGGGATATCCAGTAGTAAAAGGCAGATTGTACACGCGTTACGCACCCGTACGCCGGTCTAGGAGGCCGAAGCCCCTTGCCCCTCGACTTGCATGTTTTAGGCCTGCCGCTAGCGTTCATC
+
FFF/1BFFFFBFBF1ECFFFFF1B/BAACCFAFB8/ABB:CFFFFFF-9EFFFFFFFFFFFFBA,8//898F-.@,,=?8@@B,:88:9.BB@@BB8,,@=-8:.88B@@@@..:>@BB@.B@BBBB@B:@@.CC.:/:/BA?@A.::.CCBB>B.:8B-.9.@:,:BB@.@..8::CC.:.@@,8?>??,./,.::.!:;B,0,!090B.8,8?<.,97,8.?=,.,,7+,9FEE@F?CHHHEFEE?FEF1/?FFEFFEFFFFEEEFBFHHFCFFFB2FABF?FECFCCHFFFFHHHHHFFF
Thanks a lot for any help!
Niccolo`

Jose Navas
Niccolo`
--
---
You received this message because you are subscribed to the Google Groups "Qiime Forum" group.
To unsubscribe from this group and stop receiving emails from it, send an email to qiime-forum...@googlegroups.com.
For more options, visit https://groups.google.com/groups/opt_out.
Jose Navas

Tony Walters
Niccolo`
--
Niccolo`
thanks for your replies!
as you both suggested I tried to solve my problem changing the -r -p and -q parameters: I decreased -p until 0.1 and increased -r until 20 but still all my sequences were removed. Then I tried to decrease -q, and I was finally able to keep most of the sequences using the default (-q 3), but when I look at the histogram file most of the sequences are too short (only 70 bp)(that is the point where quality drops)!
split_libraries_fastq.py -i stitched.fastq --sample_id Y -o split_libraries/ -m Ymapfileforsplitlibr.txt --barcode_type 'not-barcoded' -p 0.1 -r 20
I am still surprised that I cannot use this file because the R1 and R2 files were not quite good in term of quality (just as usual going down at the end of the reads).
The alternative way I have been using is combining Mothur and Qiime: I run my R1 and R2 files in Mothur using the make.contigs script to stitch them, remove reads with ambiguous bases, merging duplicates sequences and removing chimeras using Uchime; then I move to Qiime for OTU picking and the following analyses. How does this approach look to you?
My main concern is that I cannot run split_libraries.py on the file I can create in this way since make.contigs in Mothur gives back only a fasta file, so I don`t have a real quality check of my sequences and I don`t know if the taxonomic assignment and the following analyses are reliable..
Do you know any way that I can use to overcome this problem? or the only other chance I have is use R1 and R2 files separately?
Any help would be really apprecited!
Thanks
Niccolo`

Brenda Koster
Niccolo`

Mike
Niccolo`
thanks for your reply and the info regarding Pandaseq! It is really interesting what they say!
I usually use first Cutadapt to trim the primers from my reads (R1 and R2) and then try to stitch the 2 files. So you can try to use Cutadapt to not depend on Pandaseq for data containing both primers.
I tried to use FLASH instead of Pandaseq: using default parameters, the program is giving me as output an extended.frags file only with very few reads, so apparently it combines only very few reads...do you have any idea why? Do you know if there is something I have to change to make it work properly?
Thanks
Niccolo`
Mike
-r 250 -f 180 -s 10 -m 15 -M 280 - x 0.35
If I remember correctly I had assembled ~93% or more of my reads. I'd have to get back to you as I am currently at the ASM meeting. I plan on assembling a bunch of Illumina data within the next month. I should be able to provide more detail on my work flow then.
-Mike
Niccolo`
thanks for the information!
I tried with the following parameters
-r 230 -f 300 -s 30 -m 15 -M 160 (my fragment length is around 300 bp and my reads are around 230bp)
but the program gives me this message:
"Read length has to be in the range (0,170]. If the reads are longer than 170, please change the constant value for READ_LENGTH in utilities.h"
Then I changed the first line in the file utilities.h from #define READ_LENGTH 170 to #define READ_LENGTH 250
Since I can see that you also used -r above 170, could you please help me to solve this problem?
Thanks a lot
Niccolo`
Mike
Niccolo`
oh I see! I was not really able to spot the link to the latest version (but you`re right, it was not so obvious!)
With the latest version, everything was fine! :)
Thanks you very much
Cheers
Niccolo`
Mike
Niccolo`
so I think FLASH worked very well!
I used -m 10 (it gave me same results of -m 15) and tried different values for -x (0.05, 0.1 and 0.2, so from a very conservative approach to a less conservative one) (I decide to stay quite low with the value for this parameter, since from the comparison with other merging scripts it is usually suggested to use low values...), and I was able to merge from 79 to 93% of my reads respectively.
After that I ran my data through split_libraries_fastq.py with -q 20 and default values of -r and -p, and I was able to keep around 90% or more of my reads, keeping also the size of the fragments I am interested in.
I would say I am quite happy with these results so far...are they similar to yours?
Cheers
Niccolo`
Mike
-Mike

saltyk
I am attempting to use FLASH to assemble R1 & R2 files generated by a MiSeq. After running FLASH, I filtered my index files to remove the reads that were eliminated by FLASH and then checked to make sure that my new stitched read and index files had the same number of sequences. All appeared to work well until I brought the files back into QIIME and then got the following error when I ran split_libraries.py:
qiime.split_libraries_fastq.FastqParseError: Headers of barcode and read do not match. Can't continue. Confirm that the barcode fastq and read fastq that you are passing match one another.
Sample Index file header:
@M00459:4:000000000-A191G:1:1101:13176:2189 1:N:0:7
NGATAAGC
+
#>>AAA4B
@M00459:4:000000000-A191G:1:1101:16100:2197 1:N:0:7
GGATAAGC
+
AAABAFDD
Sample assembled read header:
@M00459:4:000000000-A191G:1:1101:13176:2189 1:N:0:7
TACGTAGGGGGCGAGCGTTGTCCGGAATCACTGGGCGTAAANAGCGTGTAGGCNNNTNAGTCAGTCTGGTGTGAAATCTCGGGGCTCAACCCCGAGCGGTCACTGGATACTGCTGGGCTAGAATCCGGAAGAGGGGAGTGGAATTACTGGTGTAGCGGTGGAATGCGCAGATATCAGGAGGAACACCAATGGCGAAGGCAGCTCGCTGNNNCGGTATTGACGCTGAGACGCGAAAGCGTGGGGAGCGAACAGN
+
ABBA>FFBBBBBGGGGGGGGGGHGGGGGFHGHHHHHGGGGH#1BFFGGGEFHF###1#???FEHDG4?3GHHGHHEDHFH0?EDGGEFHF?DA/B////?CD#11G0DFFHFH##<CGHAGCBD#<DGBCC0C?##GFGFH#F9C#FCHH#DGEGFHHFGD/FE?/G4HHGGHGF1FHHHHGHFHFFB1GGGHGHHHHGEEA0CFFBA###EB3FHDGGGGHHHHGGEGGGGGGGGGFGGGBBBBBFBAA>>#
@M00459:4:000000000-A191G:1:1101:16100:2197 1:N:0:7
GACGTAGGGCGCGAGCGTTGTCCGGATTTATTGGGCGTAAAGAGCTCGTAGGCGNNTTGTTGCGTCGACCGTGAAAACCCGGGGCTCAACTTCGGGCTTGCGGTCGATACTGGCGGGCTGGAGTTCGGTAGGGGAGACTGGAATTCCGGGGGTAGCGGTGAAATGCGCAGATATCAGGAGGAACACCGGTGGCGAAGGCGGGTCTCTGGGCCGATACTGACGCTGAGGAGCGAAAGCGTGGGGAGCGAACAGG
+
AABBABCAAFADAGGGGGGGGGHGG?AFHHHHHGHHEEAEHH5GEFHGGEGGHG##1BB?FCF@FG//EEEEEFFHGHEE/<CCGCGCG0GB2?DD//?CGD@:<AD.=A#D<GEC<@F#BCC9C#<G#HEFG=#HFFGF;<<99BF#BF#GCC/E?EGBHEDEEEF4HFFF33E2HFGGFE>/>CDFGEGFEHDE>EEGCFFG1GGGE?E?HDFHFEGGGHHHHG2HEFEGFGGFAAFGGBBBBAFFBABAA
I have looked at the headers and don't see any differences. I tried running
"diff" on the stitched read and index files and got a long output that I have no idea how to interpret. It begins like:
29a30,32
> M00459:4:000000000-A191G:1:1101:12062:3737 1:N:0:7
> M00459:4:000000000-A191G:1:1101:15752:3741 1:N:0:7
> M00459:4:000000000-A191G:1:1101:13810:3742 1:N:0:7
53a57,64
> M00459:4:000000000-A191G:1:1101:19294:3810 1:N:0:7
> M00459:4:000000000-A191G:1:1101:10215:3810 1:N:0:7
> M00459:4:000000000-A191G:1:1101:13521:3813 1:N:0:7
> M00459:4:000000000-A191G:1:1101:13385:3816 1:N:0:7
> M00459:4:000000000-A191G:1:1101:20340:3827 1:N:0:7
> M00459:4:000000000-A191G:1:1101:18682:3830 1:N:0:7
> M00459:4:000000000-A191G:1:1101:19710:3834 1:N:0:7
I have looked at both the stitched read and index files and gone to lines 29 and 30 for example to see if I can spot differences but don't see any.
My understanding was that FLASH out was formatted to use directly in QIIME. Any suggestions?
Thanks
-Kristin
Mike
Kristin Saltonstall
--
---
You received this message because you are subscribed to a topic in the Google Groups "Qiime Forum" group.
To unsubscribe from this topic, visit https://groups.google.com/d/topic/qiime-forum/GKs2wyIjsFM/unsubscribe.
To unsubscribe from this group and all its topics, send an email to qiime-forum...@googlegroups.com.
Mike

{kind=link}
Doug
Mike R
-Mike