Converting from .gff to .bb (bigBed) with gff2bed and bedToBigBed

David da Silva Pires
I am trying to convert the following GFF file to bigBed format:
ftp://ftp.sanger.ac.uk/pub/project/pathogens/Schistosoma/mansoni/Latest_assembly_annotation_others/Schistosoma_mansoni_v5.2.gff
As an intermediate step, I converted the file to BED format using the gff2bed command:
gff2bed --keep-header --max-mem 8G < Schistosoma_mansoni_v5.2.gff > Schistosoma_mansoni_v5.2.gff.bed
After that, I sorted the BED file, computed the sizes of the chromosomes and tried to convert the sorted BED file to a bigBed file, using the following commands:
sort -k1,1 -k2,2n Schistosoma_mansoni_v5.2.gff.bed > Schistosoma_mansoni_v5.2.gff.sorted.bed
bedToBigBed Schistosoma_mansoni_v5.2.gff.sorted.bed schMan1.chrom.sizes Schistosoma_mansoni_v5.2.gff.bed.bb
The schMan1.chrom.sizes file contains two columns in each row: (i) the name of a chromosome and (ii) the size of this same chromosome. It was computed with the following command:
twoBitInfo schMan1.2bit stdout | sort -k2rn > schMan1.chrom.sizes
The schMan1.2bit file was obtained from the following fasta file:
ftp://ftp.sanger.ac.uk/pub/project/pathogens/Schistosoma/mansoni/Latest_assembly_annotation_others/Schistosoma_mansoni_v5.2.fa
However, the bedToBigBed command returned the following error:
column #10 isSizeLink do not match: Yours=[0] BED Standard=[1]
asObjects differ.
Does anyone know what I have to do to get the .bb file from the gff file?
Best regards.
--
David da Silva Pires

Hiram Clawson
The 'bed' file produced by gff2bed is somewhat like a bed file in
the first four columns, but then it has numerous string fields
that are not formatted for a 'bed' file. You could use this
file with bedToBigBed with an appropriate .as definition file
for those extra fields. It depends upon how you want to use
this big bed file.
A simple way to use your bed file would be to chop off the
extra columns after 4 and use it as a standard bed4 file.
In addition, the chromosome names are too long to function
in the bedToBigBed conversion. They have to be 31 characters
or less. You can transform your bed file to four columns
and shorter chrom names:
sort -k1,1 -k2,2n Schistosoma_mansoni_v5.2.gff.bed \
| sed -e 's/^Schisto_mansoni.//;' \
| grep -v "^_header" | cut -f1-4 > Schistosoma_mansoni_v5.2.gff.sorted.bed
And correspondingly, your chrom.sizes file:
sed -e 's/^Schisto_mansoni.//;' schMan1.chrom.sizes > short.chrom.sizes
Then:
bedToBigBed Schistosoma_mansoni_v5.2.gff.sorted.bed short.chrom.sizes \
Schistosoma_mansoni_v5.2.gff.bed.bb
--Hiram
David da Silva Pires
Thank you very much for answering my question.
The big bed file we are trying to get should be used as a way to visualize the intersection with other private track hubs. The GFF file which is subject to convertion to big bed file contains annotations that indicate exons, introns, 3' and 5' genome coordinates, besides the name of already annotated genes. We call this set of SMPs (Schistosoma mansoni predictions). We would like to map (using bowtie and tophat programs) our private contigs (built from RNA-Seq reads) against the genome of S. mansoni and then visualize, with the help of UCSC Genome Browser, which contigs are mapped against some SMPs.
With your help and the help of other friends from my lab, I managed to get a first view:
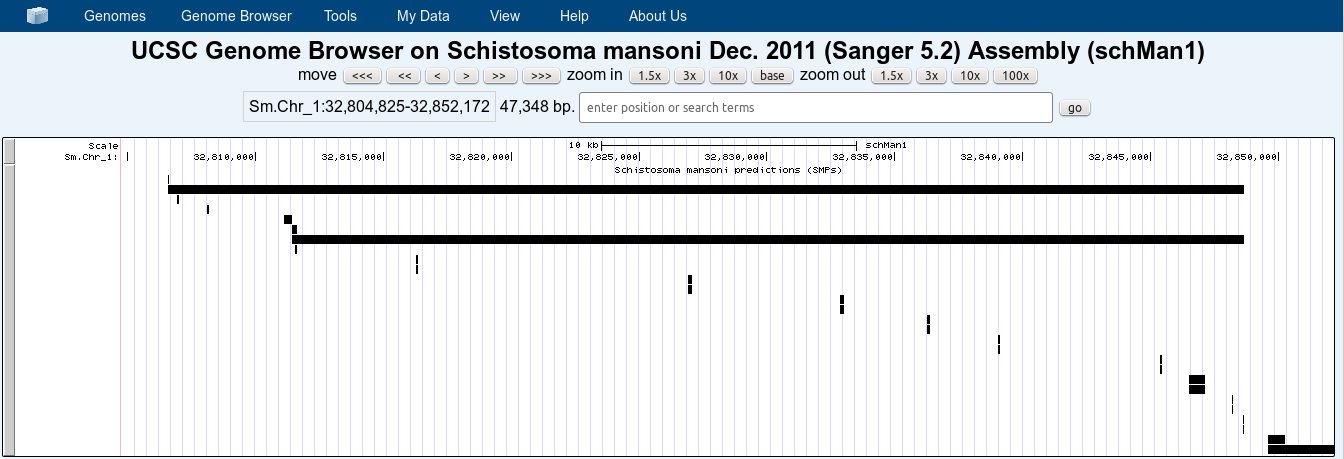
For that, we also need to change the names of the chomossomes in the genome fasta file and convert it to .2bit again, so that the mapping could be done.
The result shown was obtained from a bed file with only 6 columns. We note that, on full visualization mode, it is possible to distinguish isoforms and exons, but each one on its own line and without the gene name to the left (although the name can be checked by clicking on the exon box). It would be great if we could view exons and introns in the same line, with each one represented by lines of different thicknesses.
It seems that the problem with the bed file generated from gff2bed command is related to the use of the same character that indicates the column separator within some strings that should be considered just one column. I will try to learn how to configure the .as definition that you mentioned in order to get a bed 12 file. In parallel, another student in my lab, which already has extensive experience with the GFF file in question, is trying to write a specific parser to this GFF.
Hiram, do you know why the names of the genes were not shown next to the genes? What else I have to do to achieve this, since the names are at the fourth column?
Again, thanks for the help.
Greetings.

Jonathan Casper
Hello David,
Am I correct in thinking that you are using this bigBed file as part of a track hub? If so, please check the type specification of the bigBed file in your hub's trackDb.txt file. If the type is only listed as "type bigBed", then the bigBed file will be treated as a 3-column BED for display. That means, among other things, that gene names will not be displayed. You can fix this by adding a number to specify how many BED fields are available. Adding "type bigBed 6" to your trackDb.txt, for example, will tell the browser to display the data as a 6 column BED file (including gene name, strand, and scoring if desired).
I hope this is helpful. If you have any further questions, please reply to gen...@soe.ucsc.edu or genome...@soe.ucsc.edu. Questions sent to those addresses will be archived in publicly-accessible forums for the benefit of other users. If your question contains sensitive data, you may send it instead to genom...@soe.ucsc.edu.
--
Jonathan Casper
UCSC Genome Bioinformatics Group
--