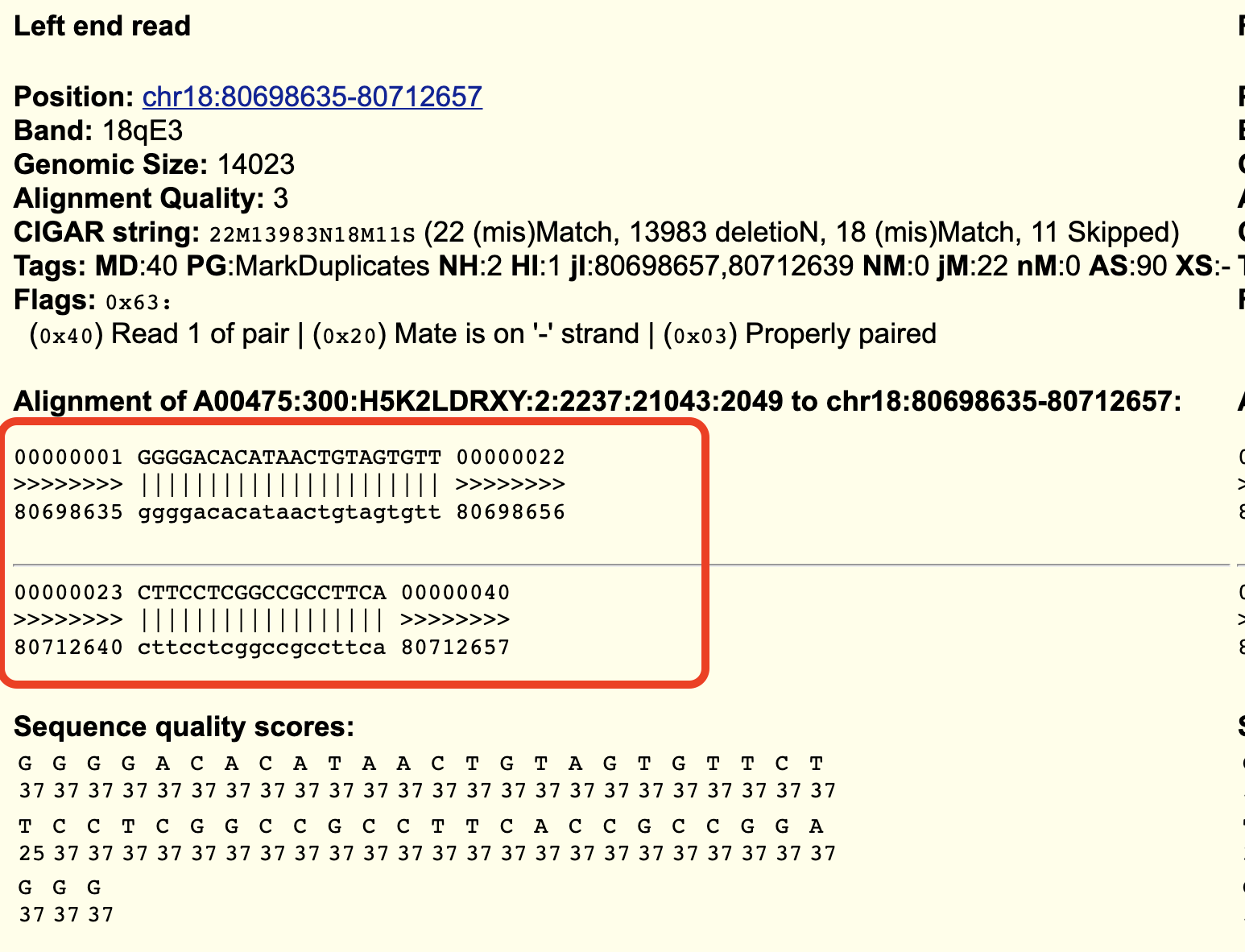
[help] BAM "Alignment of" in custom track.

Edahí Gonzalez-Avalos

Jairo Navarro Gonzalez
Hello,
Thank you for using the UCSC Genome Browser and sending your inquiry.
You can generate a display like this by using the tool, pslPretty, which will require you to convert
your BAM file into a PSL file. You can accomplish this using the bamToPsl tool. We recommend that
you filter the BAM file to the specific region that you want to visualize as it will increase the speed
of running pslPretty. To filter your BAM file to a specific position range, our engineers recommend
using samtools, http://www.htslib.org/.
You can download these tools, pslPretty and bamToPsl, from the utilities directory:
https://hgdownload.soe.ucsc.edu/downloads.html#utilities_downloads
When running pslPretty, the target.lst file corresponds to the 2bit file and query.lst are the FASTA
sequences inside the BAM file.
I hope this is helpful. If you have any further questions, please reply to gen...@soe.ucsc.edu.
All messages sent to that address are archived on a publicly accessible Google Groups forum.
If your question includes sensitive data, you may send it instead to genom...@soe.ucsc.edu.
Jairo Navarro
UCSC Genome Browser
Want to share the Browser with colleagues?
Host a workshop: https://bit.ly/ucscTraining
--
---
You received this message because you are subscribed to the Google Groups "UCSC Genome Browser Public Support" group.
To unsubscribe from this group and stop receiving emails from it, send an email to genome+un...@soe.ucsc.edu.
To view this discussion on the web visit https://groups.google.com/a/soe.ucsc.edu/d/msgid/genome/CAGGWum16gr%3DojAGiwYUEawYWwkcFoUob2GMny2-cEX6-ce%2Br4Q%40mail.gmail.com.
Edahí Gonzalez-Avalos

Brian Lee
Dear Edahi,
Thank you for using the UCSC Genome Browser and your question about using command-line tools to reproduce alignment information.
To have success with these steps we recommend that you filter the original BAM file down to the specific region that you want to process. You can use samtools to do this step to convert a sizeable bam file to a smaller bam of the region of interest:
samtools view -b -o smallerOutputBam.bam largerOriginalInput.bam "chr1:200000-300000"
To help illustrate the steps I will use an RNA-seq bam file from ENCODE on the human hg19 assembly. First I will get the underlying human hg19 assembly in the 2bit format. Then I will get the bam file in question and the index bam.bai file as well and rename the example file to make the other commands more straightforward:.
wget https://hgdownload.soe.ucsc.edu/gbdb/hg19/hg19.2bit wget http://hgdownload.soe.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeCshlLongRnaSeq/wgEncodeCshlLongRnaSeqK562CellTotalAlnRep2.bam wget http://hgdownload.soe.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeCshlLongRnaSeq/wgEncodeCshlLongRnaSeqK562CellTotalAlnRep2.bam.bai mv wgEncodeCshlLongRnaSeqK562CellTotalAlnRep2.bam exampleFile.bam mv wgEncodeCshlLongRnaSeqK562CellTotalAlnRep2.bam.bai exampleFile.bam.bai
With these files in place I will first extract a portion of the bam file into a smaller bam file. Then that smallerBam.bam will be converted to psl format. In that step a -fasta=reads.fa option will generate a fasta file that contains the reads of query sequences (reads.fa). Next the pslPretty step will create a pretty.output file and using the alignment information in psl (pslFile.psl) against the target sequence (hg19.2bit) and the query sequence in fasta (reads.fa).
samtools view -b -o smallerBam.bam exampleFile.bam "chr1:934210-934285" bamToPsl -fasta=reads.fa smallerBam.bam pslFile.psl pslPretty pslFile.psl hg19.2bit reads.fa pretty.out
You can compare this with loading the bam file on the Browser and clicking into the item. Here is a session of that file loaded as a custom track: http://genome.ucsc.edu/s/brianlee/prettyPslCheck
If you scroll down to the highlighted read you can click into the item and on the details page you will see this:
Alignment of PAN:3:93:1671:402/2/1 to chr1:934210-934285: 000001 CCGGCGGACGCCGCCAGCGACCTNCAGACAGTTGCCCTGGGAGGAGGGCG 000050 >>>>>> ||| | |||||| ||||| |||| ||||| | ||||||||||||||||| >>>>>> 934210 ccgcccgacgcccccagccacctgcagaccgcggccctgggaggagggcg 934259 000051 GGACCCCGGCGCGGCGTGGCTGCGGG 000076 >>>>>> |||||||||||||||||||||||||| >>>>>> 934260 ggaccccggcgcggcgtggctgcggg 934285
This will be similar to the information in the pretty.out file for this named item (PAN:3:93:1671:402/2/1):
$ cat pretty.out | grep -A 7 PAN:3:93:1671 >PAN:3:93:1671:402/2/1:0-76 of 76 chr1:934209+934285 of 249250621 CCGGCGGACGCCGCCAGCGACCTNCAGACAGTTGCCCTGGGAGGAGGGCGGGACCCCGGC ||| | |||||| ||||| |||| ||||| | ||||||||||||||||||||||||||| ccgcccgacgcccccAGCCACCTGCAGACCGCGGCCCTGGGAGGAGGGCGGGACCCCGGC GCGGCGTGGCTGCGGG |||||||||||||||| GCGGCGTGGCTGCGGG
Thank you again for your inquiry and for using the UCSC Genome Browser. If you have any further public questions, please send new questions to gen...@soe.ucsc.edu. All messages sent to that address are archived on a publicly accessible forum to help others find answers to similar questions. If your question includes sensitive data, you may send it instead to genom...@soe.ucsc.edu, which is a private internal list to our support team.
All the best,
To view this discussion on the web visit https://groups.google.com/a/soe.ucsc.edu/d/msgid/genome/CAGGWum29r2B6EfcpNWPxnBtv%2B5Rzh59G%3D4O9-HFXox2o%3D4OV6w%40mail.gmail.com.