Downloading Ensembl canonical transcripts with Gene IDs

Hossein Asgharian

{kind=link}
Cath Tyner
Hello Hossein,
At this time, we don't have a way to add the transcript or gene ID to the Table Browser sequence output.
However, it may be possible for you to achieve this through scripting.
The knownCanonical table does include an ENSG id, where ENSG id is the "protein" column.
Using the Table Browser, you could output all of the hg38.knownCanonical ENSG IDs along with the associated regions.
You could then get other IDs from related fields, based on the ENSG ID. Finally, you could do some scripting to match
the regions in your sequence output such that you add in the IDs that you would like to appear in the header.
Generally, scripting advice is outside the scope of this mailing list, but I do have some scripting steps that should work for you.
You might want to try the steps below with a smaller region such as chr1 and then do the same steps for the genome.
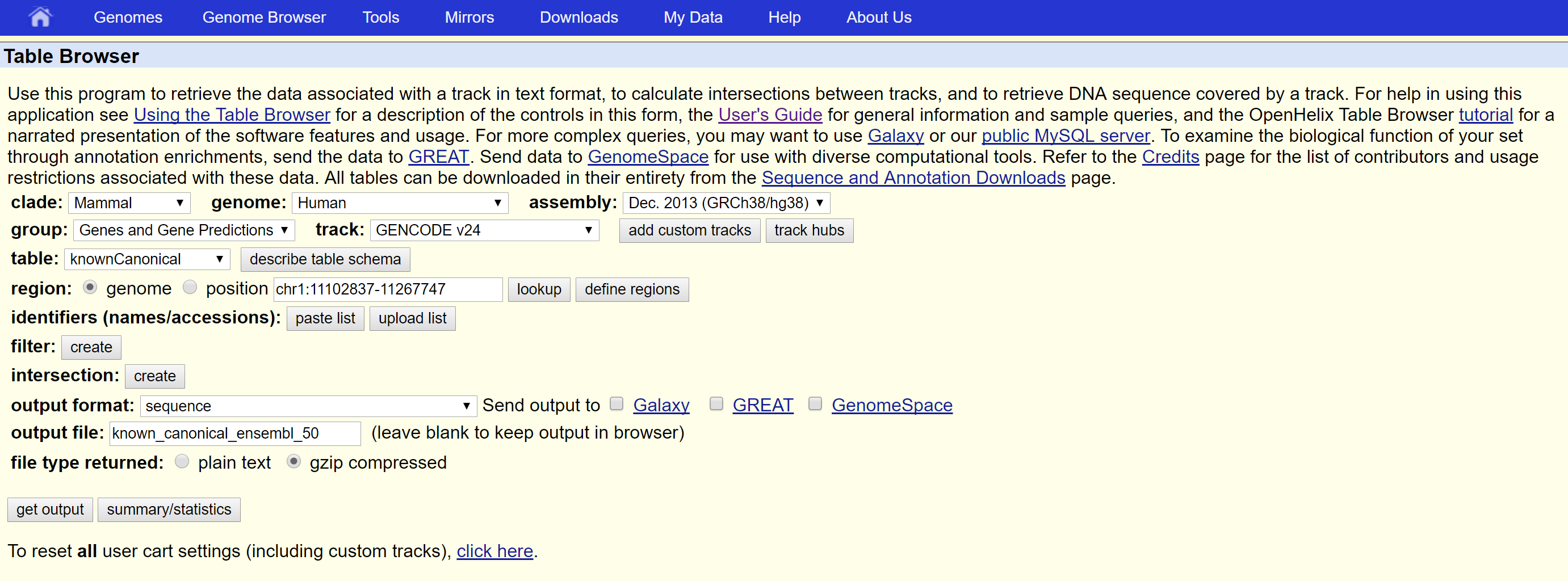
With the same Table Browser settings you have in your screenshot,
(changing from 'genome' to 'chr1' if you want a smaller example test):
1. Change the output format to "selected fields from primary and related tables".
2. Click "get output" to go to the next step.
3. Select the following checkboxes:
hg38.knownCanonical
chrom
chromStart
chromEnd
transcript (UCSC ID)
protein (ENSG ID)
hg38.kgXref fields
geneSymbol
Linked Tables
knownToEnsembl
4. Click 'allow selection from checked tables' (at the bottom of the form) to go to the next step.
hg38.knownToEnsembl
value (ENST)
5. Click 'get output'
This will give you output like this:
We now have 2 files, one is the sequence file that you already have (I'll call this file "knownCanonical.sequence") where you would like to add the transcript/gene names.
The other file is the one we just created from the Table Browser, which includes all of the related IDs that you would like (I'll call this file "knownCanonical.IDs"). The common
values in both of these 2 files are the regions, so we can use these as a mapping key value after some massaging.
The coords in each file are in different formats (0-start BED vs 1-start positional), which are described here:
http://genome.ucsc.edu/blog/the-ucsc-genome-browser-coordinate-counting-systems/
The knownCanonical.sequence file has regions in the positional format (note the start coord):
This same region in the "knownCanonical.IDs" file is in BED format (note the off-by-one start region):
Let's convert all the regions in the "knownCanonical.IDs" file from BED to positional format by adding 1 to the start.
This will give us the matching key value to make a join with the two files.
First let's look at our grep example from above and make sure the awk command (below) does what we want.
Yes, looks good, 1 was added to the start and the regions are now in positional format, thus matching the regions in the sequence file.
At this point you now have 2 files with a matching key value for positional-formatted region. You will need one final scripting step which
will replace any matching region in knownCanonical.sequence with the matched region in knownCanonical.IDs.positional, replacing it with
the entire line from the matched row in knownCanonical.IDs.positional. To do this, let's change the header in the sequence file so that it
only contains the region.
Example output, note that everything except the region was stripped from the header:
Now that we have a tidy sequence file for which to match up the key value, let's do the final step of replacing the region-only header
in the sequence file with the matching line in our knownCanonical.IDs.positional file:
Example output, you should now have a sequence file with all of the IDs that you would like:
At this point you'll want to so some thorough checking.
A quick check on my example file looks good, as I compare my original sequence file with my knownCanonical.sequence.headerWithIDs file:
Comparing the original sequence file:
as compared to the final outcome:
Also of possible interest:
http://genomewiki.ucsc.edu/index.php/Genes_in_gtf_or_gff_format
--
---
You received this message because you are subscribed to the Google Groups "UCSC Genome Browser Public Support" group.
To unsubscribe from this group and stop receiving emails from it, send an email to genome+un...@soe.ucsc.edu.
To post to this group, send email to gen...@soe.ucsc.edu.
Visit this group at https://groups.google.com/a/soe.ucsc.edu/group/genome/.
To view this discussion on the web visit https://groups.google.com/a/soe.ucsc.edu/d/msgid/genome/CAFZPC-%2BD_yrLowDo_BYJt3TcTc-1g3wf_s99Uq30HsJgxS56gQ%40mail.gmail.com.
For more options, visit https://groups.google.com/a/soe.ucsc.edu/d/optout.