Bond order between Rgroup and Rsite ?

mederich
Hi everybody,
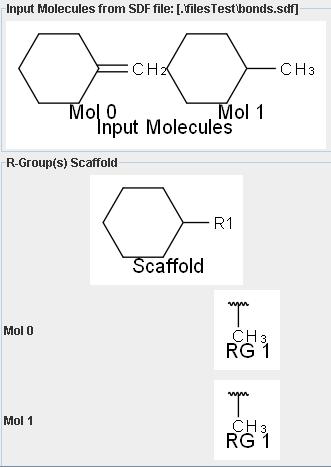
I would like to know if it’s possible to retrieve the bond order between Rgroup Attachment Point and his corresponding Rsite atom on Scaffold to re-assemble molecule with correct bonds.
For the moment I think it’s not possible (show attached image).
I hope you will find a way to fix it.
With best regards.
Médérich
Médérich Besnard
Ariana Pharma
28, rue du Dr Finlay - 75015 Paris - France
Tel: +33 (0)1 44 37 17 05 - Fax: +33 (0)1 44 37 17 01

Savelyev Alexander
Finally, I have got time to finish and resolve all the issues with
RGroup Decomposer (in this and other threads). The given issue is
related to the MOL format restriction. You can not save an attachment
point bond order. But other formats , e.g CML seems for convenient
for such a task (attachment point is considered as a simple atom,
thus, you can specify an order to attachment point). So for CML
output, I think we will add the attachment points order support, so
you can easily restore the initial molecules.
Also, for molfiles and smiles, another solution can be applied: I am
going to add an option, where the result RGroup attachment points will
be saved as a separate atom (Any atom) with a mapping (this solution
will be applied for molfiles also). The SMILES format actually keeps
AP in this way, e.g. R1=N,R2=O will be saved as R1 = *N, R2 =*O. As I
have noticed from the Gerhard's letter, he saves the R-Group number
for attachment points. The question here: do you need an additional
mapping, say, for the given example it would be: R1 = [*:1]N, R2 =[*:
2]O ?
With best regards,
Alexander
GGA Software Services LLC
Savelyev Alexander
For the CML format: bond orders will be supported for attachment points
(see rg_example_cml.jpg).
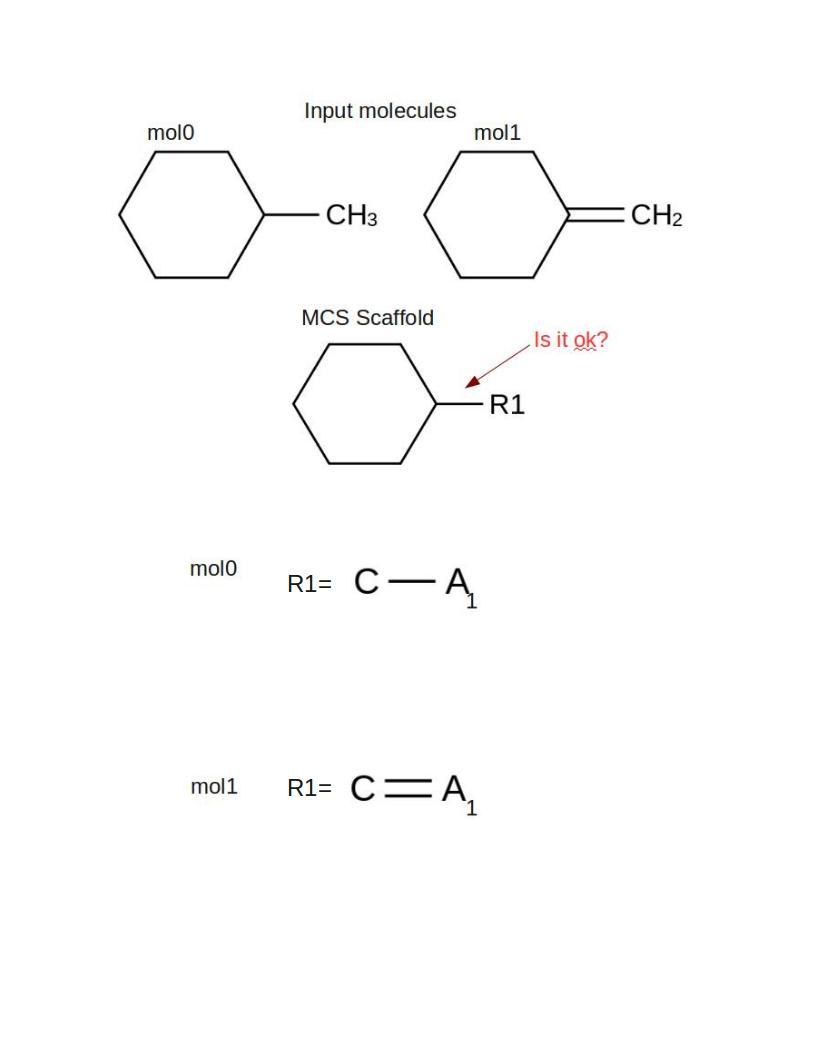
As you can see, there are bond orders specified in RGroups attachment points. I have a question for the scaffold RSite in this case: is it ok to set all the bond orders between scaffold and Rsites by single bonds. There is an option to set such bonds as 'ANY' bonds.
Also, as I mentioned before, I am going to add an option to save attachment points as any atoms with mapping (see rg_example_option.jpg). In this case, mapping will respond to attachment point number. In this example, user can re-assemble molecules with correct bonds. Such a format can be saved in molfile. The question about scaffold RSite bond order remains. If you have any questions, or, corrections for the format to be more convenient (even if it is an useless feature, in your opinion), please let us know
Best regards,
Alexander

{kind=link}
{kind=link}
{kind=link}
mederich...@gmail.com
First:
Yes is ok if you set all the bond orders between scaffold and Rsites
by single bonds. The most importante that I can get the bond order
between AP and neighbor atom in Rgroup to re-build molecule.
Second:
Your 2 proposals seem to be good (CML format and your option to save
AP as any atoms with mapping).
With best regards.
Médérich
On 20 fév, 14:56, Savelyev Alexander <asavel...@ggasoftware.com>
wrote:
> 44KAfficherTélécharger
>
> rg_example_option.jpg
> 39KAfficherTélécharger
Savelyev Alexander
Copy the correspond answers from other threads:
The new 'deco-save-ap-bond-orders' option was added. Within the option
output molecule RGroup attachment points are saved as pseudo atoms
(named 'AP1', 'AP2' etc). Therefore, the option allows to save initial
bond orders.
Retrieving bond order with 'deco-save-ap-bond-orders' option is very
simple. You just need to iterate over atoms in a RGroup and search
pseudo atoms with the 'AP' substring. The following python script (the
similar on java and c#) prints all attachment point bond orders .
--------------------------------------------------------------------------------------------
indigo = Indigo()
indigo.setOption("deco-save-ap-bond-orders", True)
mols = []
for smiles in ["OC1CCCCC1", "O=C1CCCCC1"]:
mol = indigo.loadMolecule(smiles)
mols.append(mol)
scaffold = indigo.loadQueryMolecule("C1CCCCC1")
deco = indigo.decomposeMolecules(scaffold, mols)
for item in deco.iterateDecomposedMolecules():
# get molecule with rgroups
print ("MOLECULE # %d" % item.index())
mol = item.decomposedMoleculeWithRGroups()
# iterate rgroups
for rg in mol.iterateRGroups():
print("RGROUP # %d" % (rg.index()))
if rg.iterateRGroupFragments().hasNext():
# get rgroup molecule
rg_next = rg.iterateRGroupFragments().next()
# print rgroup molfile
print(rg_next.molfile())
# iterate over rgroup atoms
for atom in rg_next.iterateAtoms():
# search for AP pseudo symbol
if(atom.symbol().find("AP") >= 0):
# iterate over atom neighbors
for atom_nei in atom.iterateNeighbors():
# get neighbor bond
bond = atom_nei.bond()
# print bord order (1 = single, 2 = double, 3 =
triple, 4 = aromatic)
print("BOND ORDER = %d" % bond.bondOrder())
With best regards,
Alexander
> Dear Alexander,
>
> First:
> Yes is ok if you set all the bond orders between scaffold and Rsites
> by single bonds. The most importante that I can get the bond order
> between AP and neighbor atom in Rgroup to re-build molecule.
>
> Second:
> Your 2 proposals seem to be good (CML format and your option to save
> AP as any atoms with mapping).
>
> With best regards.
>
> M�d�rich
>
> On 20 f�v, 14:56, Savelyev Alexander<asavel...@ggasoftware.com>
> wrote:
>> I have added examples for the future options to be more understandable.
>> For the CML format: bond orders will be supported for attachment points
>> (see rg_example_cml.jpg).
>> As you can see, there are bond orders specified in RGroups attachment
>> points. I have a question for the scaffold RSite in this case: is it ok to
>> set all the bond orders between scaffold and Rsites by single bonds. There
>> is an option to set such bonds as 'ANY' bonds.
>> Also, as I mentioned before, I am going to add an option to save attachment
>> points as any atoms with mapping (see rg_example_option.jpg). In this case,
>> mapping will respond to attachment point number. In this example, user can
>> re-assemble molecules with correct bonds. Such a format can be saved in
>> molfile. The question about scaffold RSite bond order remains. If you have
>> any questions, or, corrections for the format to be more convenient (even
>> if it is an useless feature, in your opinion), please let us know
>>
>> Best regards,
>> Alexander
>>
>> rg_example_cml.jpg
>> 44KAfficherT�l�charger
>>
>> rg_example_option.jpg
>> 39KAfficherT�l�charger
mederich...@gmail.com
I have used your new option to retrieve Rgroup AP bond order.
It's work but I have some problems when I want to export Rgroups.
In my algorithm I have to generate Rgroup molfile string using
molfile() function.
However I obtain an exception ("molecule: getImplicitH() does not work
on pseudo-atoms") on the following Rgroup:
c(:[*]):c:c:c:c:[*] |$;AP1;;;;;AP1$|
Moreover I have seen that all previous functions like
countAttachmentpoint(), iterateAttachmentPoint or clearAttachmentPoint
don't work anymore.
Do you have plan to fix them?
I hope you can help me.
Best regards.
Médérich.
On 13 avr, 13:08, Savelyev Alexander <asavel...@ggasoftware.com>
wrote:
Savelyev Alexander
The bug with an aromatic bond connected to a pseudo atom while saving
molfiles was fixed. The fixed version will be available in the upcoming
indigo rc release.
You are right. The attachment points functions do not work since the
option only saves attachment points as additional pseudo atoms.
Therefore, user have to handle those atoms manually. But we are going to
implement following features in the nearest future:
- fix all functions for attachment points
- add correct attachment bond orders loading from molfiles and SMILES
- fix attachment points rendering.
- add new API for getting attachment point bond order
I should notice that 'AP' prefix will be probably replaced by the '_AP'
prefix (chemaxon SMILES extension for attachment points). Thus, it will
be possible to load molfiles using external tools and save to the
supported SMILES format.
With best regards,
Alexander
> Hi Alexander,
>
> I have used your new option to retrieve Rgroup AP bond order.
> It's work but I have some problems when I want to export Rgroups.
>
> In my algorithm I have to generate Rgroup molfile string using
> molfile() function.
> However I obtain an exception ("molecule: getImplicitH() does not work
> on pseudo-atoms") on the following Rgroup:
> c(:[*]):c:c:c:c:[*] |$;AP1;;;;;AP1$|
>
> Moreover I have seen that all previous functions like
> countAttachmentpoint(), iterateAttachmentPoint or clearAttachmentPoint
> don't work anymore.
> Do you have plan to fix them?
>
> I hope you can help me.
> Best regards.
>
> M�d�rich.
mederich...@gmail.com
As usual I thank you for your quick answer.
When new features to handle AP pseudoAtoms will be impemented could
you share some exemple?
Best regards.
Médérich
On 25 avr, 15:40, Savelyev Alexander <asavel...@ggasoftware.com>
wrote:
Savelyev Alexander
Yes, of course, I will provide examples.
With best regards,
Alexander
>
> As usual I thank you for your quick answer.
>
> When new features to handle AP pseudoAtoms will be impemented could
> you share some exemple?
>
> Best regards.
>