Initial guess of the density matrix

Simone Piccinin
I'm studying a large molecule containing 4 Ru atoms. These are
arranged as to form two pairs. I'd like to study the molecule in a
singlet state, and see if the coupling within each pair is
ferromagnetic (FM) or antiferromagnetic (AF). Is there a way in
quickstep to guide the scf cycle toward the FM or AF solution, for
example through an appropriate initial guess of the density matrix?
Thanks for your help.
Best wishes,
Simone Piccinin
CNR-IOM Democritos (Trieste)

Jörg Saßmannshausen
I am currently tackling a similar problem but with a different TM. My approach
to the problem is to run a broken symmetry single point calculation. So I fix
the spin of each TM (say up and down), write out the individual MOs and then
calculate the overall molecule by assembling the individual MOs.
However, I am not using cp2k for that. I too would be interested if that can
be done with cp2k (broken symmetry).
Not a solution, but maybe a direction where to go?
Regards
J�rg
> --
> You received this message because you are subscribed to the Google Groups
> "cp2k" group. To post to this group, send email to cp...@googlegroups.com.
> To unsubscribe from this group, send email to
> cp2k+uns...@googlegroups.com. For more options, visit this group at
> http://groups.google.com/group/cp2k?hl=en.
--
*************************************************************
J�rg Sa�mannshausen
Research Fellow
University of Strathclyde
Department of Pure and Applied Chemistry
295 Cathedral St.
Glasgow
G1 1XL
email: jorg.sassm...@strath.ac.uk
web: http://sassy.formativ.net
Please avoid sending me Word or PowerPoint attachments.
See http://www.gnu.org/philosophy/no-word-attachments.html

marci
something in the direction of a biased initial guess is already
present in QS.
The ATOMIC guess is normally based on the construction of a first
density matrix
by occupation of atomic orbitals according to the electronic
configuration
of each element.
However, there is the possibility to modify this procedure by
subtracting or adding
valence electrons (NEL) from specific channels (identified by N and
L ) of selected elements.
This can be done independently for spin up (ALPHA) and spin down
(BETA).
Have a look at the reference inputs in cp2k/tests/QS/regtest-bs.
This might provide a better initial guess, also for charged systems.
As a safety check, if the resulting density matrix amounts to the
wrong number of electrons,
it is rescaled by a proper factor, and the calculation proceeds.
This trick is only used for the initial guess, no further constraints
are applied afterwards.
Hence, there is no guarantee that the selected configuration is kept
during the SCF.
Kind regards
Marcella
Simone Piccinin
thanks a lot for the answer.
I'm not sure I understand how to use these feature, though.
The relevant section of the cu2cl6_m1_clp6cud9.inp file in the test/QS/
regtest-bs folder is this:
&KIND Cu1
ELEMENT Cu
BASIS_SET DZVP-MOLOPT-SR-GTH
POTENTIAL GTH-BLYP-q11
&BS
&ALPHA
NEL -1 0
L 0 2
N 4 3
&END
&BETA
NEL -1 -2
L 0 2
N 4 3
&END
&END
&END
Does this mean that we are removing, from the alpha channel of Cu1, 1
electron from the 4s and in the beta channel 1 electron from the 4s
and 2 electrons from the 3d?
If so, Cu1 now has 4 electrons less than the neutral atom. A similar
treatment is done for Cu2. For Cl, on the other hand, it seems that we
are adding 2 alpha
electrons and 2 beta electrons in the 3p orbitals. Since we have 6 Cl
and 2 Cu atoms in the molecule, the overall charge should be 2*4-6*4 =
-16, resulting
in a wrong number of electrons. Is this what is done, or did I
misunderstand something?
Thanks a lot.
Best wishes,
Simone
marci
Hi
you are almost right. Actually what the keywords say in this case is
that for Cu1 1 el. 4s is taken away in spin up, while 1 4s and 2 3d
are taken in spin down. The misunderstanding comes from the fact that
cp2k is working for each spin independently, always using as a
reference the full electronic configuration, and if LSD is active a
factor 2 is applied afterwards.
Then Cu is 4s-1 3d-10. The change imposed for the bias leads to Cu1
ALPHA 4s-0 3d-10 . After the factor 2 is 4s-0 3d-5.
Cu1 BETA instead becomes 4s-0 3d-4. At the end Cu1 has one unpaired
spin-up.
It should become clearer by reading the extended output that appears
with print level MEDIUM.
Example
##########
Spin 1
Guess for atomic kind: Cu1
Electronic structure
Total number of core
electrons 18.00
Total number of valence
electrons 10.00
Total number of
electrons 28.00
Multiplicity not
specified
S [ 2.00 2.00 2.00]
P [ 6.00 6.00]
D 10.00
#########
Indeed, for this system the counting of electrons after the bias must
be readjusted, because each spin channel turns out to have one
electron too much. In the output you can read:
#######
Re-scaling the density matrix to get the right number of electrons for
spin 1
# Electrons Trace(P)
Scaling factor
33
33.999 0.971
######
and later on
######
Re-scaling the density matrix to get the right number of electrons for
spin 2
# Electrons Trace(P)
Scaling factor
33
33.999 0.971
#####
Hope this helps,
Marcella
flo
actually there is a second way, which could solve your problem, in
case the intial guess is not sufficient.
CP2K provides a section WFN_MIX in QS%PRINT. This keyword can be used
to reorder the MOs in the restart file.
It has to be used as follows:
You need to run a complete wavefunction optimization, and specify:
RESULT_MO_INDEX as the index of the MO to be modified (you can specify
the spin of this MO as well) and
ORIG_MO_INDEX to be the MO from which the WFN to be pasted originates
from.
If you want for example to swap the alpha and beta spins of two states
(1 and 2) you will have to specify
&WFN_MIX
&UPDATE
RESULT_MO_INDEX 1
RESULT_SPIN_INDEX ALPHA
RESULT_SCALE 0.000000000
ORIG_MO_INDEX 2
ORIG_SPIN_INDEX BETA
ORIG_SCALE 1.0000000000
ORIG_IS_VIRTUAL .FALSE.
&END
&UPDATE
RESULT_MO_INDEX 2
RESULT_SPIN_INDEX BETA
RESULT_SCALE 0.0000000
ORIG_MO_INDEX 1
ORIG_SPIN_INDEX ALPHA
ORIG_SCALE 1.0000000
ORIG_IS_VIRTUAL .FALSE.
&END
&END
like this the beta WFN of MO 2 is moved to alpha MO 1 and alpha MO1 is
moved to beta MO 2.
If you now do a restart using the RESTART_WFN (the modified WFN simply
replaces the original RESTART_WFN, therefore, the restart file won't
have a special name), the inital guess will have the state specified
above.
I don't know if this really fits your problem, but as far as I could
see, it should work.
regards
Flo
Don't know if this solution
Simone Piccinin
thanks a lot, now I think I understand how to use this option.
If this doesn't solve my problem I'll try Flo's suggestion.
Best wishes,
Simone
marci
On Feb 11, 9:12 pm, Simone Piccinin <piccinin.sim...@gmail.com> wrote:

tao liu
I can understand the &BS setting for Cu1, but still confused on Cl and Cl1.
&KIND Cl
BASIS_SET DZVP-MOLOPT-GTH
POTENTIAL GTH-BLYP-q7
&BS
&ALPHA
NEL 2
L 1
N 3
&END
&BETA
NEL 2
L 1
N 3
&END
&END
&END
&KIND Cl1
ELEMENT Cl
BASIS_SET DZVP-MOLOPT-GTH
POTENTIAL GTH-BLYP-q7
&BS
&ALPHA
NEL 2
L 1
N 3
&END
&BETA
NEL 0
L 1
N 3
&END
&END
&END
As suggested by Marcella,
for 'Cl' in the input file, Alpha is (5+2)/2=3.5, and same for Beta, in the end 'Cl' should have 3s-2 3p-7
for 'Cl1' in the input file, Alpha is (5+2)/2=3.5, and (5+0)/2 =2.5 for Beta, in the end 'Cl1' should have 3s-2 3p-6
Could anyone tell me if I understand correctly or not ?
Thanks,
Tao

S Ling
--
You received this message because you are subscribed to the Google Groups "cp2k" group.
To unsubscribe from this group and stop receiving emails from it, send an email to cp2k+uns...@googlegroups.com.
To post to this group, send email to cp...@googlegroups.com.
Visit this group at https://groups.google.com/group/cp2k.
For more options, visit https://groups.google.com/d/optout.

katarína stančiaková
I am trying to set up calculation for my Cu+ system, however, even after reading this thread I cannot understand how the electrons are assigned to channels in CP2K. I wanna test two possible spin states (just for testing purposes) of Cu+:
Cu+ with electron configuration 3d9 4s1 (Cu1) and 3d10 4s0 (Cu2). I tried to build my input sections:
&KIND Cu1
ELEMENT Cu
BASIS_SET DZVP-MOLOPT-GTH
POTENTIAL GTH-BLYP-q11
&BS
&ALPHA
&ALPHA
NEL 0 0
L 0 2
N 4 3
&END
&BETA
NEL -1 -2
L 0 2
N 4 3
&END
&END
&KIND Cu2
ELEMENT Cu
BASIS_SET DZVP-MOLOPT-GTH
POTENTIAL GTH-BLYP-q11
&BS
&ALPHA
&ALPHA
NEL -1 0
L 0 2
N 4 3
&END
&BETA
L 0 2
N 4 3
&END
Is this correct?
Katarina
Dňa streda, 10. februára 2016 11:53:26 UTC+1 S Ling napísal(-a):

Krack Matthias (PSI)
Dear Katarina
You may try for Cu1 alpha
NEL 1 0
L 0 2
N 4 3
instead of
NEL 0 0
L 0 2
N 4 3
Hope that works.
Matthias
katarína stančiaková
Krack Matthias (PSI)
Dear Katarina
you have indeed to approach each spin channel in the BS section from a “closed shell” point of view. The slides 22 and 23 from the last CP2K tutorial in Paderborn show an example for a BS section for UO2. The BS section is not perfectly intuitive.
HTH

jiaj...@gmail.com
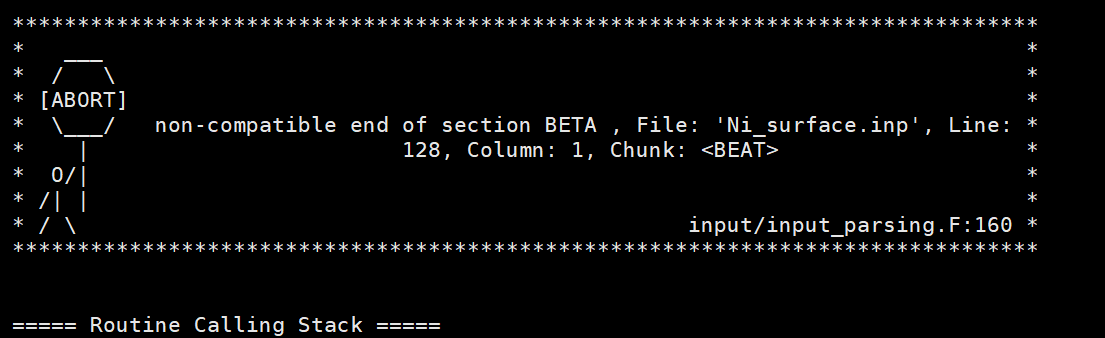
在 2018年11月8日星期四 UTC+8下午11:37:18,Matthias Krack写道:

Martin Brehm
as the error message already suggested, you just have a typo in your input file:
"&END BEAT"
:-)
Best regards,
Martin